
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Cronkhite-Canada syndrome with hypothyroidism: Clinical, endoscopic and pathologic pitfalls
Dapeng Wang; Christopher McPhaul; Debby Rampisela*
Department of Pathology, Baylor Scott & White Medical Center, Temple, TX 76508, USA.
*Corresponding Author : Debby Rampisela
Department of Pathology, Baylor Scott & White Medical Center, Temple, TX 76508, USA.
Email: Debby.Rampisela@BSWHealth.org
Received : Apr 16, 2021
Accepted : May 17, 2021
Published : May 20, 2021
Archived : www.jcimcr.org
Copyright : © Rampisela D (2021).
Abstract
Cronkhite-Canada Syndrome (CCS) is a rare non-hereditary protein-losing enteropathy, and characterized by ectodermal and gastrointestinal abnormalities. The etiology remains unclear, however, some studies suggest the likelihood of immune dysregulation. The diagnosis of CCS is challenging because the clinical symptoms are non-specific and the endoscopic and pathologic findings can overlap with other processes. This can potentially lead to delay in diagnosis, resulting in delayed patient management. We reported a 74-year-old male presented with initial symptoms of abdominal pain and nausea, and later with diarrhea, weight loss, hypothyroidism and multiple strokes. Gastric Antral Vascular Ectasia (GAVE) was suspected on the initial endoscopy. The diagnosis of CCS was rendered on the third biopsy. After steroid treatment, the patient’s clinical symptoms were relieved. In this paper, we discussed the clinical, endoscopic and pathologic pitfalls of CCS.
Citation: Wang D, McPhaul C, Rampisela D. Cronkhite-Canada syndrome with hypothyroidism: Clinical, endoscopic and pathologic pitfalls. J Clin Images Med Case Rep. 2021; 2(3): 1152.
Introduction
CCS was first described in 1955 by Leonhard Wolsey Cronkhite and Wilma Jeanne Canada in two women with diarrhea, gastrointestinal polyposis, pigmentation of the skin, alopecia and atrophy of the fingernails and toenails [1]. Since then more than 500 cases have been reported worldwide, most cases were from Asian countries, mainly Japan [2]. The syndrome primarily affects adults with a slight male predominance and peak incidence between 50 and 60 years old [3]. The characteristic features include triad ectodermal abnormalities (dystrophic finger and toe nails, hyperpigmentation and alopecia) along with gastrointestinal abnormalities (diarrhea, malabsorption and polyposis). These polyps can develop in any sites of gastrointestinal tract, but more commonly seen in the stomach and the colon. The most common initial symptom of the reported cases is diarrhea; however, one paper from Japan stated that the most common initial symptom was taste disorder/hypogeusia [4]. The classic endoscopic and pathologic finding for CCS is edema with polyposis, but cases have been documented without polyposis [3,5]. The variable clinical, endoscopic and pathologic findings make the diagnosis of CCS challenging. Moreover, the etiology of CCS is obscure. One proposed theory is that immune dysregulation is the underlying cause of the syndrome [6,7]. Once diagnosed treatment typically involves corticosteroids along with support therapy. Early diagnosis is critical because mortality in CCS is reported to be greater than 50% [8].
Case report
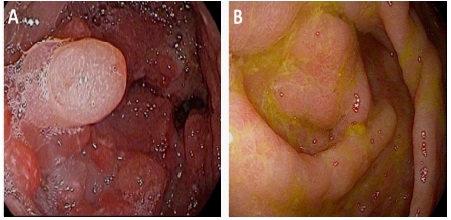
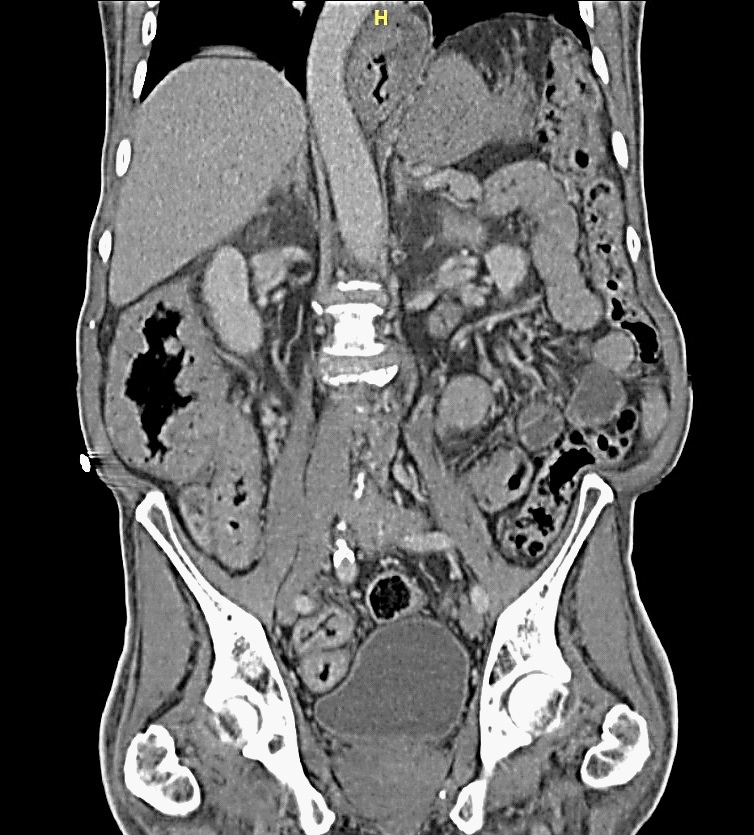
A 74-year-old Caucasian man presented to our GI clinic for abdominal pain, nausea, bloating and 45 lbs weight loss in 6 months. Four months prior he had presented to a surgeon’s office at an outside facility and underwent an esophagogastroduodenoscopy/EGD, which revealed a thickened, nodular, erythematous antrum that was biopsied. There was a concern for malignancy so he underwent a CT scan and PET scan of the abdomen and pelvis. Both of these studies revealed a thickened distal stomach but no lymphadenopathy, or increased uptake to suggest malignancy. His biopsy results was reported to show hyperplasia and no evidence of malignancy. A repeat EGD also showed thickened nodular erythematous antrum. He was given the diagnosis of Gastric Antral Vascular Ectasia (GAVE). The patient continued to lose weight with decreased oral intake, constant nausea and persistent non-bloody diarrhea. In the middle of the workup of all the above symptoms, he was diagnosed with hypothyroidism and had been started on levothyroxine. In our institution repeat EGD and colonoscopy (Figure 1) were performed, showing diffuse mucosal abnormalities, involving the stomach (thickened edematous folds with nodularity), small bowel (thickened edematous folds with scalloping), and colon (thickened edematous folds with polypoid lesions) with rectal sparing. The esophagus was unremarkable. CT abdomen (Figure 2) showed circumferential wall thickening of the ascending colon, cecum, and terminal ileum. Multiple laboratory tests were done. Tests for infections and celiac disease were negative. The abnormal findings were as follows: Positive anti-nuclear antibody/ANA (speckled 1:320), high C-reactive protein (28.7 mg/L), slightly high TSH (4.62 mcIU/mL), slightly lower total T3 (0.38 ng/mL) and free T3 (2.1 pg/mL), and slightly lower hemoglobin (11.9 gm/dL), RBC (3.95 x10 e12/L), hematocrit (36.6 %), calcium (7.6 mg/dL), protein total (5.1 gm/dL) and albumin (2.1 gm/dL). The differential showed slight increase of monocytes (13%). During the work up in our institution, the patient developed significant weakness and required a walker. He also had a presyncopal episode and multiple strokes. Brain MRI showed multiple punctate areas of restricted diffusion within the cerebellar vermis and left cerebellar hemisphere consistent with recent, likely embolic, infarctions. Later the biopsies from the stomach, small bowel and colon showed findings consistent with CCS (see histopathologic findings below for more detail findings). More careful physical exam demonstrated onychodystrophy of finger and toe nails, and cutaneous hyperpigmentation of palms and macular lesion of the chest consistent with CCS. The patient was started on prednisone, ranitidine, parenteral nutritional therapy and went to a rehabilitation facility. His abdominal symptoms resolved and his strength improved. The last follow up two months later showed resolution of symptoms. A follow up endoscopy was scheduled, but a few days after his last follow up he passed away for unclear reason.
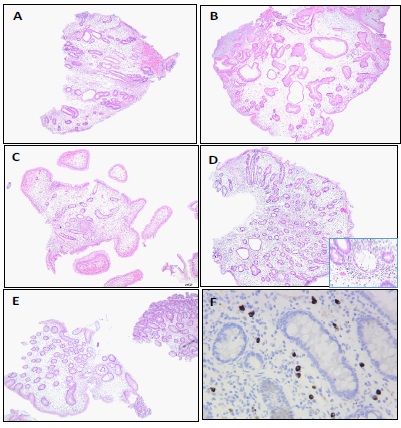
Histopathologic findings (Figure 3)
Duodenal biopsy showed focal active duodenitis with marked lamina propria edema and subtotal villous blunting. Stomach biopsy showed inflammatory type polyp with marked lamina propria edema and no evidence of Helicobacter pylori organisms. Terminal ileum biopsy showed focal active enteritis with marked lamina propria edema. Jejunum and ileum biopsies showed focal active enteritis with marked lamina propria edema and subtotal villous blunting. Colon biopsy showed low-grade tubular adenoma with marked lamina propria edema, and separate fragments of colonic mucosa with focal active colitis and marked lamina propria edema. IgG4 immunostains were done on all specimens and showed increased number of IgG4 positive plasma cells (up to 20 per high power field).
Discussion
There was a delay in CCS diagnosis for our patient, this was likely due to the non-specific initial symptoms, the unawareness of CCS entity because of its rarity, and the unfamiliarly with the physical examination, endoscopic and histologic findings of CCS. There was also associated hypothyroidism in this patient which adding the confusion. However, due to unresponsiveness after treatment, repeat endoscopy and biopsies led to the diagnosis of CCS. The patient also had multiple strokes, which were likely thromboembolic complication of the CCS as he did not have any noted thromboembolic predisposition prior to his initial symptoms of CCS. In this patient the clinical symptoms did resolve after treatment, however, the patient later passed away. The cause of death was not clear but is may related to stroke due to incompliance with anticoagulant treatment.
The majority of the reported CCS patients have an initial presentation with diarrhea, weight loss, abdominal pain, and nausea-vomiting. Other presentations included dysgeusia/hypogeusia, tinnitus, hematochezia/melena, constipation, paresthesia, xerostomia, alopecia, nail changes, upper limb weakness and pain [2-4,7,9,10]. The ectodermal changes (dystrophic nail, hyperpigmentation and alopecia) usually follow the other clinical symptoms by a few weeks or months, however, there are cases reported with nail changes and/or hair loss preceding other symptoms [2,10]. Varieties of clinical diagnoses are often considered in these patients including hypothyroidism, Crohn’s disease, celiac disease, infection and occult malignancy [5,11-15]. CCS patients have also been reported to have numerous concurrent diagnoses; including vitiligo, systemic lupus erythematosus, scleroderma, rheumatoid arthritis, membranous nephropathy, hypothyroidism, myxedema, myopathy and gastric outlet obstruction syndrome [4,8,9,11-14,16-18]. The pathogenesis for these concurrent diagnoses is unclear.
The most common endoscopic finding of CCS is diffuse mucosal edema with polyposis. Other reported endoscopic findings include celiac disease-like appearance with blunting of the duodenal villi, colitis-like appearance with colonic mucosal erythema, loss of vascularity and ulceration, Gastric Antral Vascular Ectasia (GAVE) syndrome-like appearance with mosaic pattern of the stomach and giant hypertrophic gastritis/Menetrier’s disease-like appearance with diffusely enlarged gastric mucosa folds [5,18-20]. Polyps in CCS can be seen in any part of the intestine, however, they are much less likely to be seen in the esophagus. Only two studies reported the presence of polyps in the esophagus [18,21]. The stomach and colon were reported to have more severe involvement by polyps compared to the small intestine and rectum, and for the small intestine duodenum had more polyps compared to the jejunum and ileum [20].
The radiographic findings of CCS show polypoid filling defects or diffuse coarsening of the mucosal pattern [20-23]. Kilcheski et al suggested that the radiographic appearance of the stomach with enlarged rugal folds, polyps, whiskering (appearance of multiple tiny projections due to barium trapped between nodular excrescences of rugae) and hypersecretion were characteristic radiologic appearance of the stomach in CCS [22].
Just like the endoscopic finding, the most common histopathologic finding of CCS is diffuse edema with polyposis. Other pathologic findings include enlarged gastric and small intestinal folds, villous atrophy, enteritis, colitis, collagen deposition, non-caseating granulomas and lymphangiectasia and adenocarcinoma [20]. The initial histopathologic diagnoses include eosinophilic gastroenteritis, Menetrier’s disease, Helicobacter gastritis, duodenitis, celiac disease, enteritis, colitis, colitis cystica, inflammatory fibroid polyp and descriptive diagnosis [5]. The pathological hallmark of CCS has been the universal mucosal edema and expansion of the lamina propria, and there is also atrophic mucosa in at least a portion of the gastrointestinal tract. Multiple biopsies from different sites of the gastrointestinal tract are critical for diagnosing CCS. The finding of diffuse mucosal edema in different sites of gastrointestinal tract is very important because there have been a few reported cases with absence of polyposis [3,5,21,24]. The polyps in CCS includes hyperplastic polyp, tubular adenomas, juvenile/hamartomatous polyp and inflammatory polyp. It is worth noting that the polyp and non-polypoid areas both typically show edema. Electron microscopic examination in one reported study showed obscured gastric pit, distorted colonic crypts, irregular size of surface epithelial cells of the gastric and colonic mucosa, and conspicuous mucoid/mucous granules [25]. The risk of neoplasia is controversial, although the current weight of evidence points to an increased risk of adenomas and carcinomas [1]. The majority of malignancies were reported in the stomach, distal colon and rectum. The incidence of gastric and colorectal cancer in patients with CCS is approximately 10-20% [16,18]. The polyposis slowly regresses, indicating that long term monitoring likely needed to assess treatment outcomes, even, in most cases, after remission of clinical symptoms. Given the slow regression of the polyposis and the likelihood of increased risk of neoplasia, a continued/annual endoscopic surveillance even after clinical remission is suggested [18]. The etiology of CCS is unclear, however, an autoimmune cause is suspected. Elevated autoimmune markers including antinuclear antibodies, IgG plasma cell infiltrate of the gastrointestinal biopsies, association with other autoimmune diseases like vitiligo, systemic lupus erythematosus, scleroderma, hypothyroidism and membranous glomerulonephritis, and clinical improvement with immunosuppressive therapy support an autoimmune etiology [7,24,26,27].
The complication of CCS are related to the size, location and morphology of the polyps and the results of massive fluid, protein and electrolyte losses. The complications include gastrointestinal bleeding, protein-losing enteropathy, malnutrition, vitamin deficiencies, anemia, intussusception, rectal prolapse, thromboembolic episodes, congestive heart failure and coagulation abnormalities [16,18,20]. Thromboembolic episodes are likely attributed to dehydration, congestive heart failure or coagulation abnormalities [20].
Death related to CCS is usually secondary to bronchopneumonia, cachexia, anemia, septicemia or congestive heart failure [15,29]. The mainstays of medical treatment for CCS are steroid therapy and nutritional support [18,20]. Other treatments include cyclosporine A, octreotide, anti-TNFalpha agent, azathioprine, cyclosporine, antibiotic, histamine-receptor antagonists and surgical treatment [7,18,28,29]. If Helicobacter pylori infection is present, the Helicobacter pylori should be eradicated. There were reported CCS cases with Helicobacter pylori infection, which showed remission after Helicobacter pylori eradication using antibiotics [8,30]. Therefore, it is possible that Helicobacter pylori infection is involved in the development or exacerbation of CCS polyps, but the underlying mechanisms remains uncertain.
Conclusion
CCS is a rare disease and is usually presented with non-specific initial symptoms. A thorough history and physical exam are essential for reaching the diagnosis. The presence of diffuse edema particularly with polyposis on the endoscopy should strongly alert the clinicians for CCS, and the same thing for the pathologists when the diffuse edema with polyposis seen on the biopsies. The diagnosis of CCS is based on history, physical exam, endoscopy findings and histology. Early diagnosis of CCS is important to provide appropriate treatments and follow up.
Acknowledgement: We thank Dr. Thomas P. Please from Cleveland Clinic in Cleveland, Ohio for his expert consultation for this case.
References
- Cronkhite LW, Canada WJ. Generalized gastrointestinal polyposis; an unusual syndrome of polyposis, pigmentation, alopecia and onychotrophia. N Engl J Med. 1955; 252: 1011-1015.
- Rubio CA, Björk J. Cronkhite-Canada syndrome - A Case report. Anticancer Res. 2016; 36: 4215-4217.
- De Petris G, Chen L, Pasha SF, Ruff KC. Cronkhite-Canada syndrome diagnosis in the absence of gastrointestinal polyps: A case report. Int J Surg Pathol. 2013; 21: 627-631.
- Goto A. Cronkhite-Canada syndrome: Epidemiological study of 110 cases reported in Japan. Nihon Geka Hokan Arch Jpn Chir. 1995; 64: 3-14.
- Bettington M, Brown IS, Kumarasinghe MP, de Boer B, Bettington A, Rosty C. The challenging diagnosis of Cronkhite-Canada syndrome in the upper gastrointestinal tract: A series of 7 cases with clinical follow-up. Am J Surg Pathol. 2014; 38: 215-223.
- Lin HJ, Tsai YT, Lee SD, et al. The Cronkhite-Canada syndrome with focus on immunity and infection. Report of a case. J Clin Gastroenterol. 1987; 9: 568-570.
- Sweetser S, Ahlquist DA, Osborn NK, et al. Clinicopathologic features and treatment outcomes in Cronkhite-Canada syndrome: support for autoimmunity. Dig Dis Sci. 2012; 57: 496-502.
- Kopáčová M, Urban O, Cyrany J, et al. Cronkhite-Canada syndrome: Review of the literature. Gastroenterol Res Pract. 2013; 2013: 856873.
- She Q, Jiang J-X, Si X-M, Tian X-Y, Shi R-H, Zhang G-X. A severe course of Cronkhite-Canada syndrome and the review of clinical features and therapy in 49 Chinese patients. Turk J Gastroenterol Off J Turk Soc Gastroenterol. 2013; 24: 277-285.
- Burnell RH. Cronkhite-Canada syndrome. Med J Aust. 1976; 1: 347-348.
- Størset O, Todnem K, Waldum HL, Burhol PG, Kearney MS. A patient with Cronkhite-Canada syndrome, myxedema and muscle atrophy. Acta Med Scand. 1979; 205: 343-346.
- Jones AF, Paone DB. Canada-Cronkhite syndrome in an 82-year-old woman. Am J Med. 1984; 77: 555-557.
- Pal A, Sen S, Ghosh S, Sarkar R, Jalan KN. Cronkhite-Canada syndrome with hypothyroidism. Indian J Gastroenterol. 1990; 9: 229-230.
- Qiao M, Lei Z, Nai-Zhong H, Jian-Ming X. Cronkhite-Canada syndrome with hypothyroidism. South Med J. 2005; 98: 575-576.
- Freeman K, Anthony PP, Miller DS, Warin AP. Cronkhite Canada syndrome: A new hypothesis. Gut. 1985; 26: 531-536.
- Firth C, Harris LA, Smith ML, Thomas LF. A Case Report of Cronkhite-Canada Syndrome Complicated by Membranous Nephropathy. Case Rep Nephrol Dial. 2018; 8: 261-267.
- Onozato Y, Sasaki Y, Abe Y, et al. Cronkhite-Canada Syndrome Associated with Gastric Outlet Obstruction and Membranous Nephropathy: A Case Report and Review of the Literature. Intern Med Tokyo Jpn. 2020; 59: 2871-2877.
- Watanabe C, Komoto S, Tomita K, et al. Endoscopic and clinical evaluation of treatment and prognosis of Cronkhite-Canada syndrome: A Japanese nationwide survey. J Gastroenterol. 2016; 51: 327-336.
- Urabe S, Akazawa Y, Takeshima F. A Case of Cronkhite-Canada Syndrome with a Colitis-mimicking Endoscopic Presentation. J Crohns Colitis. 2015; 9: 1179-1180.
- Daniel ES, Ludwig SL, Lewin KJ, Ruprecht RM, Rajacich GM, Schwabe AD. The Cronkhite-Canada Syndrome. An analysis of clinical and pathologic features and therapy in 55 patients. Medicine (Baltimore). 1982; 61: 293-309.
- Wen X-H, Wang L, Wang Y-X, Qian J-M. Cronkhite-Canada syndrome: report of six cases and review of literature. World J Gastroenterol. 2014; 20: 7518-7522.
- Kilcheski T, Kressel HY, Laufer I, Rogers D. The radiographic appearance of the stomach in Cronkhite-Canada syndrome. Radiology. 1981; 141: 57-60.
- Harned RK, Buck JL, Sobin LH. The hamartomatous polyposis syndromes: clinical and radiologic features. AJR Am J Roentgenol. 1995; 164: 565-571.
- Zhao R, Huang M, Banafea O, et al. Cronkhite-Canada syndrome: a rare case report and literature review. BMC Gastroenterol. 2016; 16: 23.
- Suzuki K, Uraoka M, Funatsu T, et al. Cronkhite-Canada syndrome. A case report and analytical review of 23 other cases reported in Japan. Gastroenterol Jpn. 1979; 14: 441-449.
- Riegert-Johnson DL, Osborn N, Smyrk T, Boardman LA. Cronkhite-Canada syndrome hamartomatous polyps are infiltrated with IgG4 plasma cells. Digestion. 2007; 75: 96-97.
- Fan R-Y, Wang X-W, Xue L-J, An R, Sheng J-Q. Cronkhite-Canada syndrome polyps infiltrated with IgG4-positive plasma cells. World J Clin Cases. 2016; 4: 248-252.
- Mao EJ, Hyder SM, DeNucci TD, Fine S. A Successful Steroid-Sparing Approach in Cronkhite-Canada Syndrome. ACG Case Rep J. 2019; 6: 1-4.
- Taylor SA, Kelly J, Loomes DE. Cronkhite-Canada Syndrome: Sustained Clinical Response with Anti-TNF Therapy. Case Rep Med. 2018; 2018: 9409732.
- Kato K, Ishii Y, Mazaki T, et al. Spontaneous Regression of Polyposis following Abdominal Colectomy and Helicobacter pylori Eradication for Cronkhite-Canada Syndrome. Case Rep Gastroenterol. 2013; 7: 140-146.