
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
A rare cause of familial atrioventricular block with a novel LMNA mutation (p. Ala502Val)
Drissa Mariem; Yaakoubi Wael*; Helali Sana; Hbiba Drissa
Hopital La Rabta Adult Cardiology Service Tunis Ministry of Public Health, Tunisia.
*Corresponding Author: Yaakoubi Wael
Hopital La Rabta Adult Cardiology Service Tunis
Ministry of Public Health, Tunisia.
Email: yaakoubiwael3@gmail.com
Received : Jun 02, 2021
Accepted : Jul 12, 2021
Published : Jul 16, 2021
Archived : www.jcimcr.org
Copyright : © Wael Y (2021).
Abstract
Mutation in LMNA accounts for 10% of Dilated Cardiomyopathy (DCM). It is characterised by progressive conduction system disease, arrhythmia and systolic impairment, lamin A/C heart disease is the most malignant gene common in DCMs especially in man. It is likely to be an under-recognised cause of this cardiomyopathy. In certain clinical scenarios, particularly familial DCM with early conduction disease, the probability of finding an LMNA mutation may be quite high.
Citation: Mariem D, Wael Y, Sana H, Drissa H. A rare cause of familial atrioventricular block with a novel LMNA mutation (p. Ala502Val). J Clin Images Med Case Rep. 2021; 2(4): 1238.
Introduction
LMNA gene mutations are one of the most frequent genetic abnormalities involved in dilated cardiomyopathy (DCM) and it has been estimated that LMNA mutations cause up to 10% of familial DCM [1]. LMNA mutations are associated with cardiac abnormalities characterized by arrhythmias: sinus node dysfunction, Atrioventricular Block (AVB), atrial and ventricular arrhythmias [2]. Here, we present a Tunisian family with hereditary AVB caused by the novel LMNA mutation (p. Ala502Val) documented in one member of it and described its features.
Case presentation
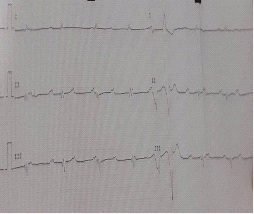
A 44-year-old man presented in clinic with dyspnea dizziness and orthopnea for 2 weeks. He denied chest pain or fever. His past medical history was unremarkable, without skeletal muscle disease, premature ageing, or metabolic disorders. In his family history we found that: His mother had a complete AVB at age of 50 years treated by double chamber pacemaker, his brother had complete AVB at age of 45 years treated by double chamber pacemaker, his sister had Mobitz I AVB at age of 45 years not yet stimulated, his Maternal uncle had complete AVB at early age treated by double chamber pacemaker, his Maternal Grandmother had complete AVB at age of 50 years treated by double chamber pacemaker. On examination, pulse rate was of 35 beats/min, blood pressure of 100/60 mmHg. There was no sign of heart failure. The ECG (Figure 1) revealed sinus rhythm with 2:1 Atrioventricular Block (AVB) alternating with complete AVB.
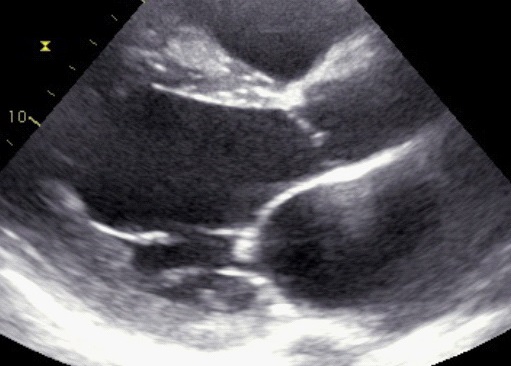
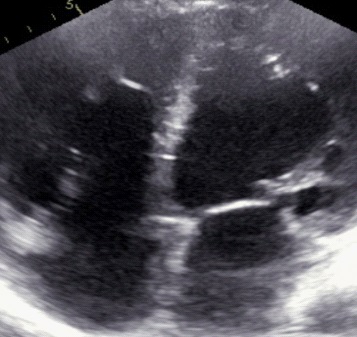
Trans-thoracic Echocardiogram (Figure 2 & 3) showed dilated left ventricle with ejection fraction estimated at 15% and global hypokinesia. The right ventricle was also dilated.
Coronary artery angiography showed no abnormality in origin, course, or atherosclerotic change. We decided to implant a triple chamber pacemaker and performed genetic testing using the TruSight Cardio Panel (Illumina, Sophia Genetics), which includes 109 genes. All associated mutations/variants were confirmed by direct Sanger sequencing. A novel LMNA mutation c.1505C>T, g.1561066920C>T (p. Ala502Val) in the LMNA gene was identified.
Due to lack of funds the rest of the family members were only explored by an TTE who did not reveal any LV dysfunction in them at exception of his brother who’s was about 38%.
He was discharged after 30 days in relatively good clinical conditions after implantation on standard pharmacological therapy for heart failure. Six months after pacemaker implantation the patient remained asymptomatic. Left ventricular dimension and function were unchanged.
Discussion
In the present case, we discovered from targeted resequencing of genes of our patient a novel LMNA mutation (p. Ala502Val) in a Tunisian family with hereditary CCD (Cardiac Conduction Disorders) and DCM.
In addition, we found intra-familial clinical gender differences in clinical severity.
Dilated cardiomyopathy is one of the most common causes of heart failure and sudden cardiac death in young people [3]. LMNA is one of the most common causal genes of CCD and DCM [4,5]. Different genetic mutations may result in different onset ages and severity of dilated cardiomyopathy. Lamins A and C, encoded by LMNA gene, are nuclear intermediate filament proteins that form one of the major structural components of the lamina network, which underlies and mechanically supports the nuclear envelope. Mutations in LMNA are responsible for more than ten different disorders, commonly called “Laminopathies”. These diseases affect tissues in a specific (striated muscle, adipose tissue, peripheral nerve) or in a systemic manner (premature ageing syndromes). Cardiac involvement is called cardiolaminopathy [6]. During our case study, we observed a gender difference in the family studied in which affected males experienced an earlier onset of CCD and a more severe DCM phenotype compared to affected females [7]. Although laminopathies have been considered monogenic, they exhibit a remarkable degree of clinical variability in severity, penetrance, and age at onset. Clinical variability has been observed among family members with the same LMNA mutation; however, the cause of the intra-familial clinical variability has remained unclear [7]. A recent study revealed significant gender differences in cardiac phenotypes such as higher mortality, and more severe cardiac dysfunction in males with DCM carrying an LMNA mutation [8]. It has been suggested that gonadal hormones may explain the gender difference found in cardiac phenotypes. Results from in vitro and in vivo studies indicate that estrogen may play a pivotal role [9,10].
Genetic testing for dilated cardiomyopathy is complicated by marked genetic heterogeneity. Today there are at least 35 different genes in which mutations have been reported to cause dilated cardiomyopathies. New genes with mutations resulting in familial dilated cardiomyopathy will undoubtedly be recognized [11]. Although the likelihood of clinically detectable LMNA-related DCM in pediatric cases is low, genetic testing should be offered as it can facilitate identification of at-risk children who may benefit from early treatment, potentially forestalling the development of advanced disease [12] .
Differences in perspective exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis
While most centers would consider decisions regarding prenatal testing to be the choice of the parents, discussion of these issues is appropriate [13].
Conclusion
Our observation illustrates the case of familial AVB associated with DCM which is related to LMNA mutation. Laminopathies are associated with a wide spectrum of disease phenotypes, ranging from skeletal muscle disease, pre-mature ageing, metabolic disorders, and cardiac abnormalities.
References
- Jacob KN, Garg A. Laminopathies: Multisystem dystrophy syndromes. Mol Genet Metab. 2006; 87: 289-302.
- Worman HJ, Bonne G. “Laminopathies”: A wide spectrum of human diseases. Exp Cell Res. 2007; 313: 2121-2133.
- Cowan JR, Kinnamon DD, Morales A, Salyer L, Nickerson DA, Hershberger RE. Multigenic disease and bilineal inheritance in dilated cardiomyopathy is illustrated in non-segregating LMNA pedigrees. Circ Genomic Precis Med. 2018; 11: e002038.
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999; 341: 1715-1724.
- Bécane HM, Bonne G, Varnous S, Muchir A, Ortega V, Hammouda EH, et al. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin Electrophysiol PACE. 2000; 23: 1661‑1666.
- Bertrand AT, Chikhaoui K, Ben Yaou R, Bonne G. Laminopathies : un seul gène, de nombreuses pathologies. Biol. 2011; 205: 147-162.
- Kawakami H, Ogimoto A, Tokunaga N, Nishimura K, Kawakami H, et al. A Novel Truncating LMNA Mutation in Patients with Cardiac Conduction Disorders and Dilated Cardiomyopathy. Int Heart J. 2018; 59: 531-541.
- van Rijsingen IAW, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013; 15: 376-384.
- Du X-J. Gender modulates cardiac phenotype development in genetically modified mice. Cardiovasc Res. 2004; 10.
- Babiker FA, De Windt LJ, van Eickels M, Grohe C, Meyer R, Doevendans PA. Estrogenic hormone action in the heart: regulatory network and function. Cardiovasc Res. 2002; 53: 709‑719.
- Judge DP. Use of Genetics in the Clinical Evaluation of Cardiomyopathy. JAMA. 2009; 302: 2471.
- Rampersaud E, Siegfried JD, Norton N, Li D, Martin E, Hershberger RE. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog Pediatr Cardiol. 2011; 31: 39-47.
- Hershberger RE, Morales A. LMNA-Related Dilated Cardiomyopathy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, et al., éditeurs. GeneReviews® . Seattle (WA): University of Washington, Seattle; 1993.