
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Cowden syndrome and Lhermitte - Duclos disease: A case report and review of the literature
Chun Peng Goh1,3; Bingcheng Wu2; Ting Ting Feng1 ; Ning Chou3
1 Division of Neurosurgery, Ng Teng Fong General Hospital, Singapore.
2 Department of Pathology, National University Hospital, Singapore.
3 Division of Neurosurgery, National University Hospital, Singapore.
*Corresponding Author: Ting Ting Feng
Division of Neurosurgery, Department of General
Surgery, Ng Teng Fong General Hospital Tower B Level
2, 1 Jurong East Street 21, Singapore 609606.
Email: tingting.feng@mohh.com.sg
Received : Jun 18, 2021
Accepted : Jul 27, 2021
Published : Jul 30, 2021
Archived : www.jcimcr.org
Copyright : © Feng TT (2021).
Abstract
Cowden syndrome (CS) is a rare, autosomal dominant, multisystem disease related to the mutation of Phosphatase and tensin homolog (PTEN) tumour suppressor gene. It is characterized by the occurrence of multiple hamartomas, mucocutaneous lesions, and is associated with a high risk of malignancies. Lhermitte–Duclos Disease (LDD), or dysplastic cerebellar gangliocytoma, is a rare hamartomatous lesion of the cerebellar cortex with a unique “Tiger Stripe” appearance on Magnetic Resonance Imaging (MRI). Since 1991, LDD has been considered pathognomonic and part of CS. In addition, as almost all adult onset LDD cases were associated with PTEN gene mutations, LDD and CS are both included in PTEN Harmartoma Tumour Syndrome (PHTS).
In this article, we report a 48-year-old female patient, who presented with a right cerebellar lesion resulting in hydrocephalus. The histology of the cerebellar lesion confirmed the diagnosis of LDD, and her clinical history is highly suggestive of CS.
Due to the high incidence of multisystemic malignancies and other disease, it is important for clinicians to be aware of the association between LDD and CS. Affected patients should be evaluated carefully and screened for cancers accordingly, so as to allow early diagnosis and treatment.
Abbreviations: CS: Cowden Syndrome; LDD: Lhermitte-Duclos Disease; PTEN: Phosphatase and tensin homolog; PHTS: PTEN Hamartoma Tumour Syndrome.
Citation: Goh CP, Wu B, Feng TT, Chou N. Cowden syndrome and Lhermitte-Duclos disease: A case report and review of the literature. J Clin Images Med Case Rep. 2021; 2(4): 1250.
Introduction
Lhermitte–Duclos Disease (LDD), also known as dysplastic gangliocytoma of cerebellum, is a rare hamartomatous lesion of the cerebellar cortex first described by Lhermitte and Duclos in 1920 [1]. LDD generally follows an indolent course, but may cause mass effect in the posterior fossa and result in hydrocephalus. Cerebrospinal fluid diversion surgery and excision of the lesion may be necessary in lesions with progressive nature [2].
In 1963, Drs. Kenneth M. Lloyd and Macey Dennis named Cowden Syndrome (CS) after their patient Rachel Cowden, who had multiple mucocutaneous lesions and passed away from breast cancer when she was only 30 years old [3]. CS is a rare autosomal dominant, multisystem disease involving harmatomatous overgrowth of tissues in all embryonic layers, resulting in multiple hamartomas, mucocutaneous lesions, and a high risk of multiple systemic malignancies such as breast, thyroid, endometrial and renal cancers [3-6]. Nonmalignant manifestations of CS include meningioma, benign breast lumps (fibrocystic disease, fibroadenomas), gastrointestinal polyps, uterine fibroids, thyroid adenomas and various mucocutaneous lesions [4-8].
In 1991, Padberg et al. proposed that LDD should be considered a manifestation of Cowden syndrome as a single phacomatosis [7]. Currently, adult onset LDD is thought to be pathognomonic with CS [6,8]. Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene encoding a lipid and protein phosphatase, which plays a crucial role in the regulation of cell growth, apoptosis, and cell cycle arrest [9]. Almost all adult onset LDD cases were associated with mutations in PTEN [6,8-10]. Furthermore, germline mutations in PTEN are associated with a group of inherited syndromes of dysfunctional cell growth, collectively known as the PTEN Harmatoma Tumour Syndromes (PHTS) [9,11].
In this article, we report a 48-year-old female patient with a right cerebellar LDD, with clinical, radiological and histological findings. We also review the literature on the diagnostic criteria, as well as the screening guidelines for malignancies associated with CS.
Clinical conditions started to improve spontaneously (NIHSS 21→15).The main diagnostic hypothesis was an embolic phenomenon with spontaneous resolution and possible distal embolism in the small vessels not visible on CT angiography.
Management
Intravenous thrombolysis with recombinant tissue-type plasminogen activator was administered (IV rt-PA 0.9 mg/kg) with subsequent further neurological improvement (NIHSS 15→12).
Nonetheless, after few minutes the patient’s blood pressure dropped abruptly (ABP 90/60 mmHg); dobutamine (8 mcg/kg/ min) and norepinephrine (0.1 mcg/kg/min) were started without benefit and acute pulmonary edema due to cardiogenic shock developed. ECG was superimposable on the previous one with ST elevation in inferior leads. Echocardiography showed severe reduction in LVEF (30%). Blood sample tests recorded a significant increase in troponin values from 349 to 500,000 ng/L.
The patient was intubated and coronary angiography was performed, showing left circumflex artery thrombosis with distal embolism and slow-flow (Figure 3). 3000 IU of unfractionated heparin was administered and thrombus aspiration was performed with Export Catheter without flow improvement.
Case presentation
The patient is a 48-year-old female with past medical history of: Bilateral breast carcinoma in 2004, right thigh hemangioma in 2012, complex endometrial hyperplasia without atypia in 2016, multiple colonic hamartomatous polyps with ganglionneuromatous elements and oesophageal glycogen acanthosis in 2016. She declined genetic testing for Cowden syndrome due to financial constraints.
This patient presented with headache of 3 weeks duration. She described the headache as gradual onset, tightness at the vertex region, and worse at night especially after lying down. It had been associated with vomiting, non-vertiginous dizziness, unsteady gait and veering to right side when walking. A non-contrast Computer Tomography (CT) scan of the brain was performed, which showed a large hypodense lesion at the right cerebellum with resulting hydrocephalus. She was then referred to neurosurgery for further management.
She underwent an emergency external ventricular drain insertion. Subsequently, an Magnetic Resonance Imaging (MRI) brain with contrast (Figure 1) was performed and showed typical ‘‘tiger-striped’’ pattern of cerebellar folia in the right cerebellum.
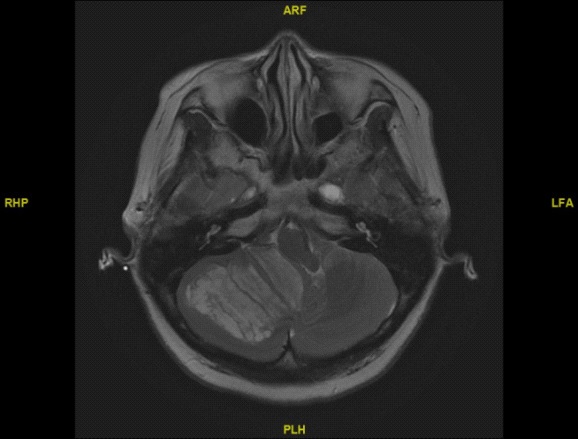
She subsequently underwent a suboccipital craniotomy and gross total resection of the tumour. Upon dura opening, hypertrophic folia was noted. The tumour was pale, soft and lobulated with good tissue plane between the brain and the tumour.
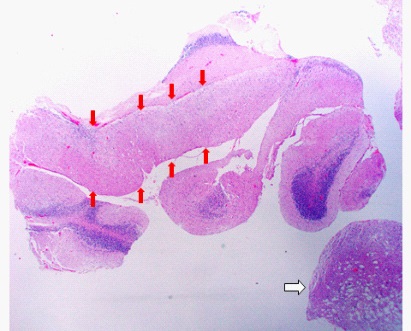
Microscopic examination revealed that the cerebellum architecture was relatively preserved. Moderate to large-sized hypertrophic ganglion cells were present in variable numbers in the molecular layer of the cerebellum with complete replacement of the internal granular layer in most areas (Figure 2). There was also extension of the hypertrophied ganglion cells into the superficial white matter. Occasional calcifications of the capillaries were noted. In addition, there was marked vacuolization and expansion of the white matter. Luxol fast stain highlighted abnormal and increased myelinization of the molecular layer, as well as a reduction in the white matter in some areas (Figures 3-6). Purkinje cells were reduced in number. There was no metastatic tumour. Immunohistochemistry revealed that the ganglion cells were positive for synaptophysin and neurofilament. They were negative for GFAP and S100. Occasional reactive astrocytes were highlighted by GFAP but there was no atypical proliferation of glial cells, excluding the diagnosis of a ganglioglioma. The overall histopathological findings were consistent with dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos Disease).
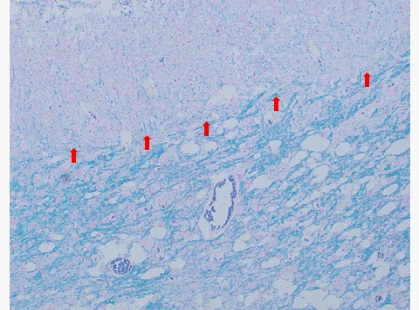
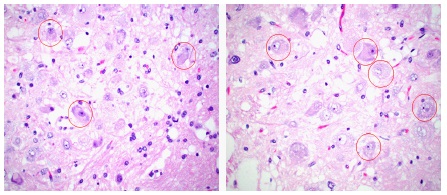

The patient subsequently had a ventriculoperitoneal shunt insertion and made full neurological recovery.
Discussion
LDD is a rare malformation of the cerebellum of uncertain etiology and pathogenesis. It was first recognized to be related to CS by Padberg et al. in 1991 [7], and is now considered to be nearly pathognomonic with CS [6,8]. It usually follows an indolent course, but the gradually increasing size of the cerebellar lesion may lead to hydrocephalus and the patient may present with clinical features of increased intracranial pressure [2]. Radiological and histopathological features of this disease are distinctive and are very rarely confused with other tumours.
Cowden syndrome is an autosomal dominant disorder characterized by multiple hamartomas and is associated with many malignancies of breast, thyroid, endometrial, kidney, colon and skin [3-5,9,10]. Germline mutations in the tumour suppressor gene PTEN at locus 10q23.2 have been identified in 1997 as the major susceptibility gene for CS [9,12]. This mutation causes the PTEN protein to have altered function leading to hyperactivity of the mammalian target of rapamycin (mTOR) pathway, which regulates cell growth, proliferation, and survival. Approximately 80% of patients with CS have a germline mutation of the PTEN gene, and another 10% harbor mutations in its promoter region [9,13]. Germline PTEN mutations are also associated with a group of syndromes under PHTS, and both LDD and CS are currently included in PHTS [8,14,15].
Diagnosis of cowden syndrome
Diagnostic criteria for Cowden syndrome was first established in 1996 before the identification of the PTEN gene and the ability to molecularly confirm a diagnosis. In 2011, Tan MH et al conducted a multicenter prospective study to select patients for PTEN mutation testing. A diagnostic criterion called Cleveland Clinic scoring system was then derived from 3042 probands satisfying relaxed CS clinical criteria. Using the Cleveland Clinic PTEN Risk Calculator, clinicians are now able to estimate an individual’s pretest probability of having a germline PTEN mutation [16]. Based on Cleveland Clinic PTEN Risk Calculator, our patient has a 99% risk of harboring a PTEN mutation.
Screening for malignancies
Cowden syndrome represents one of the several disorders in the spectrum of PHTS. Other disorders under PHTS include Bannayan-Riley-Ruvalcaba syndrome, Proteus-syndrome, and Proteus-like syndrome [6,10,14,15]. These syndromes always greatly increase the risk of malignancy due to the presence of a PTEN mutation [12]. In patients with germline PTEN mutations, lifetime risks for malignancy have been identified for breast (85%), thyroid (35%), renal (34%), and endometrial cancers (28%), with slightly elevated risks for colorectal cancers (9%) and melanoma (6%) [6]. Management guidelines for PTEN-mutation positive patients have been developed in an attempt to screen for early malignancies (Table 1) [6].
Table 1: Monitoring recommendation for Cowden related malignancies
Cancer |
Age |
Recommendation |
Female Breast |
30 |
Annual mammogram; consider MRI for dense breasts. (Consider prophylactic mastectomy) |
Thyroid |
Age of diagnosis |
Annual ultrasound |
Renal |
40 |
Imaging every two years |
Endometrial |
30 |
Annual endometrial biopsy or transvaginal ultrasound. (Consider prophylactic hysterectomy-oophorectomy unnecessary) |
Colorectal |
35-40 |
Colonoscopy with follow-up depending on results |
Melanoma |
Age of diagnosis |
Annual dermatologic exam |
Conclusion
LDD is a rare hamartomatous lesion of the cerebellar cortex, which is associated with CS. When the diagnosis of either one of these two disease is made, it is imperative to search for the other disease, due to the high risk of malignancies in patients with CS. In patients with CS or LDD, a long term follow up plan is required due to the lifetime malignancy risks. Familial screening and genetic testing is recommended as well.
References
- Lhermitte J, Duclos P. Sur un ganglioneurome diffus du cortex du cervelet. Bull Assoc Fr Etude Cancer. 1920; 9: 99–107.
- Bhatia JK, Bhatoe HS, Vadhanan S. Lhermitte-Duclos disease: A rare entity. Med J Armed Forces India. 2016; 72: S147-S149.
- Lloyd KM 2nd, Dennis M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963; 58: 136-142.
- Uppal S, Mistry D, Coatesworth AP. Cowden disease: A review. Int J Clin Pract. 2007; 61: 645-652.
- Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013; 105: 1607-1616.
- Mester J, Eng C. Cowden syndrome: recognizing and managing a not-so-rare hereditary cancer syndrome. J Surg Oncol. 2015; 111: 125-130.
- Padberg GW, Schot JD, Vielvoye GJ, Bots GT, de Beer FC. Lhermitte-Duclos disease and Cowden disease: a single phakomatosis. Ann Neurol 1991; 29: 517–523.
Jiang T, Wang J, Du J, Luo S, Liu R, et al. Lhermitte-Duclos Disease (Dysplastic Gangliocytoma of the Cerebellum) and Cowden Syndrome: Clinical Experience From a Single Institution with LongTerm Follow-Up. World Neurosurg. 2017; 104: 398-406.
Gustafson S, Zbuk KM, Scacheri C, Eng C. Cowden syndrome. Semin Oncol. 2007; 34: 428-434.
Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol. Methods. 2015; 77-78: 11-9.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006; 7: 606-619.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997 28; 275: 1943- 1947.
Arun KA, Sreejith R, Hitha B, Geetha P, Sasidharan PK. Cowden syndrome with Lhermitte- Duclos disease presenting as ataxia. Natl Med J India. 2015; 28: 74-76.
Nielson C, Fischer T, Fischer R, Donald J, Rajpara A. LhermitteDuclos Disease in association with Cowden Syndrome. Dermatol Online J. 2016; 22: 13030/qt7qn7v4bf.
Ngeow J, Eng C. Germline PTEN Mutation Analysis for PTEN Hamartoma Tumor Syndrome. Methods Mol Biol. 2016; 1388: 63-73.
Tan MH, Mester J, Peterson C, Yang Y, Chen JL, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011; 88: 42-56.