
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Sturge-weber syndrome: Characteristics of facial port wine stain with neurological and ophthalmological deficits
Prasta Bayu Putra; Retno Danarti*
Department of Dermatology and Venereology, Faculty of Medicine, Public Health, and Nursing, Universitas Gadjah Mada/ Dr. Sardjito Hospital, Yogyakarta, Indonesia.
*Corresponding Author: Retno Danarti
Department of Dermatology and Venereology,
Faculty of Medicine, Public Health, and Nursing,
Universitas Gadjah Mada/ Dr. Sardjito Hospital,
Yogyakarta, Indonesia.
Email: danarti@ugm.ac.id
Received : Aug 14, 2021
Accepted : Sep 08, 2021
Published : Sep 14, 2021
Archived : www.jcimcr.org
Copyright : © Danarti R (2021).
Abstract
Background: Sturge-Weber Syndrome (SWS) is a neurocutaneous defect involving facial Port Wine Stain (PWS) with vascular deficits in the brain and ipsilateral eyes. Facial PWS can be the initial marker and help predict the severity of SWS.
Aims and objectives: To study the association of facial PWS characteristics, neurological and ophthalmological deficits in SWS with the degree of severity.
Materials and Methods: A retrospective descriptive study of patients with SWS was conducted at Dr. Sardjito Hospital between January 2010 - December 2020. Facial PWS characteristics were assessed from photographs. Assessments of neurological and ophthalmological deficits used physical and supportive examinations, including Electroencephalogram (EEG), Magnetic Resonance Imaging (MRI), and Intelligent-Quotient (IQ).
Results: From 19 female patients aged 1 month to 26 years with facial PWS, seven fulfilled the inclusion criteria. “High risk” facial PWS V1 and P1 b distribution patterns were found in all patients. Most patients had neurological deficits of rare focal seizures (83%). Fine motor disorders (50%) were the most severe motoric deficits found. All patients experienced glaucoma with decreased visuality to blindness and hemianopsia. EEG epileptiform patterns, MRI showing angiomatosis and brain atrophy, and low IQ scores were recorded.
Conclusions: Pathogenesis and degree of severity in SWS were related to facial PWS, neurological and ophthalmological deficits.
Keywords: Sturge-weber syndrome; facial port-wine stain; neurological deficit; ophthalmological deficit; neurocutaneous disorder.
Citation: Putra PB, Danarti R. Sturge-weber syndrome: Characteristics of facial port wine stain with neurological and ophthalmological deficits. J Clin Images Med Case Rep. 2021; 2(5): 1310.
Introduction
Sturge-Weber syndrome (SWS, OMIM 185300) involves neurocutaneous defects characterized by facial capillary malformations in the form of a port-wine stain (PWS) accompanied with vascular malformations in the brain and ipsilateral eye. The incidence of SWS is estimated to range from 1 in 20,000 to 50,000 births without sex predisposition [1]. The disease is caused by somatic mutations in the guanine nucleotide-binding protein alpha-q (GNAQ) gene, which occur sporadically and not hereditarily [2]. The mutations cause hyperproliferation and apoptosis inhibition of vascular cells that lead to the vascular malformations in SWS [2].
The facial PWS is one of the SWS components which can be an initial marker and help to predict the severity of SWS (Figure 1). Classically, the distribution of PWS on the face is a “high risk” area for SWS due to the involvement of the ophthalmic nerve dermatome (V1) of the trigeminal nerve [1]. Waelchii et al. reported that the distribution of PWS according to facial embryonic vascularization (placode) is a more accurate predictor for SWS, because embryologically the vascularization of the skin, brain, and eyes are derived from the same embryonic protrusion [3]. The extent of facial PWS that occurs in patients with “high risk” area distribution may also help to predict SWS severity [4].
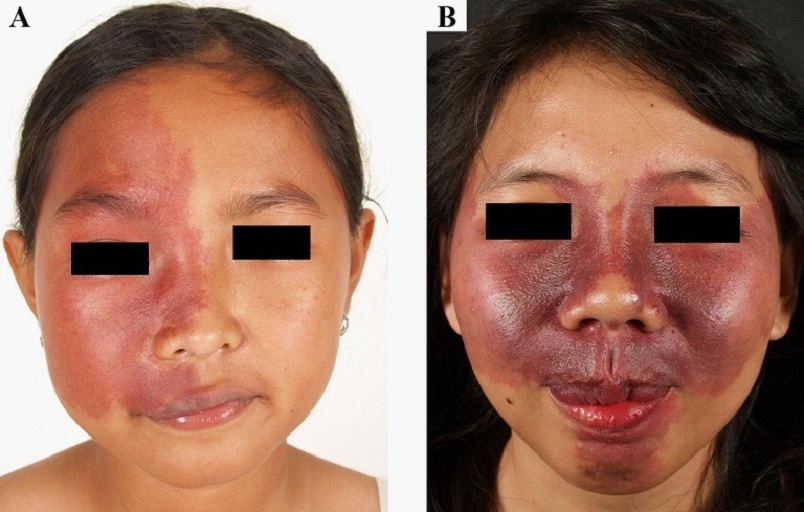
Neurological and ophthalmological deficits in individuals with SWS are progressive and occur due to vascular malformations. Neurological deficits can manifest clinically as seizures and stroke-like episodes with hemiparesis, whereas ophthalmological deficits occur as visual field defects or decreasing visuality due to glaucoma. In more severe condition, SWS can lead to cognitive disorders such as decreasing intelligence and social adaptive function. Neurological and ophthalmological diagnostic examinations need to be performed as early as possible to assist the management plan in the prevention of SWS progression [1,4].
To assess the severity of the disease, the characteristics of facial PWS, neurological, and ophthalmological deficits were examined in patients with SWS. By assessing the characteristics of facial PWS, it is expected that dermatologists can predict the possibility of complications and team up with other specialists (in ophthalmology, pediatrics, neurology, psychiatry, and psychology) to determine the most effective management of SWS.
Subjects and methods
Subjects and study design
A descriptive study was conducted with a retrospective design. The subjects were patients who were admitted to the dermatology and venereology polyclinic at Dr. Sardjito Hospital, Yogyakarta between January 2010 to December 2020. Subjects’ data were collected through medical records, clinical photographs, and diagnostic examinations. The inclusion criteria were: SWS patients with facial PWS who had been diagnosed through ophthalmological, neurological, and/or radiological examination. The exclusion criteria were: Patients with incomplete medical record data from illness or dermatological examination, and incomplete clinical photographs (complete clinical photos consist of 1 anterior side and 2 lateral sides of the face).
Clinical assessment
The characteristics of facial PWS were assessed through distribution patterns and scores based on the latest clinical photographs. The distribution pattern of facial PWS was based on the trigeminal dermatomal and facial embryonic vascularization (placode) patterns [3]. The scores of facial PWS were obtained by assessing the extent of percentage of PWS lesions in each distribution pattern, with score 0 if the area was without lesion, 1 if the extent of lesion area was 1: -25%, 2: 26-50%, 3: 51-75%, and 4: 76-100%. Scores from 6 areas of the distribution pattern were summed with a total score ranging from 0-24 for the facial PWS score [4].
Neurological deficits assessments included the type, frequency of seizures and the presence of motoric disorders. The ophthalmological deficits were measured by assessing visuality, intraocular pressure, and visual field defects. All data were taken from the latest physical and diagnostic examinations. The neurological score was assessed in subjects with complete neurological and ophthalmological deficit data (Table 1) with a total score ranging from 0-15 [4].
Table 1: Neurological score to assess the severity of the Sturge-Weber syndrome [5].
Assessment |
Score Range |
Score Description |
Field of view |
0-2 |
0: No visual field defect |
Hemiparesis |
0-4 |
0: No weaknesses |
Frequency of seizures |
0-4 |
0: none ever |
Cognitive function |
0-5 |
Infant or pre-school age Children Adult |
The latest electroencephalogram (EEG), Magnetic Resonance Imaging (MRI), and Intelligent-Quotient (IQ) data were assessed as additional data. The MRI score assessment was performed in four brain regions (frontal, parietal, temporal, and occipital) in subjects who had MRI data. Score 1 indicated no asymmetry was found, 2 for mild asymmetry (angiomatosis only), 3 for moderate asymmetry (angiomatosis and mild cerebral atrophy), and 4 indicated severe asymmetry (angiomatosis and severe cerebral atrophy). Scores from all four brain regions were summed for a total MRI score ranging from 4-16.
Ethical considerations
All subjects were asked to provide informed consent prior to the study.
Results
From January 2010 to December 2020 there were 19 SWS patients with facial PWS who were admitted to the dermatology and venereology polyclinic at Dr. Sardjito Hospital, Yogyakarta. Further searching through medical record, clinical photograph, and diagnostic examination data obtained seven patients who met the inclusion and exclusion criteria. All of the subjects were female with age range between 1 month to 26 years old. Complete data of neurological and ophthalmological deficits were found in 4 and 3 subjects, respectively. Additional diagnostic examination data of the subjects consisted of EEG, MRI, and IQ examinations (Table 2).
Table 2: Neurological score to assess the severity of the Sturge-Weber syndrome [5].
|
Gender |
Age |
Facial |
Neurological |
Ophthalmological |
EEG |
MRI |
IQ |
1 |
Female |
1 mo |
(+) |
(+) |
(±) |
(n/a) |
(n/a) |
(n/a) |
2 |
Female |
3 yr |
(+) |
(+) |
(+) |
(+) |
(n/a) |
(n/a) |
3 |
Female |
14 yr |
(+) |
(+) |
(+) |
(+) |
(+) |
(n/a) |
4 |
Female |
13 yr |
(+) |
(+) |
(+) |
(n/a) |
(n/a) |
(n/a) |
5 |
Female |
26 yr |
(+) |
(±) |
(±) |
(n/a) |
(n/a) |
(n/a) |
6 |
Female |
8 yr |
(+) |
(±) |
(+) |
(+) |
(+) |
(+) |
7 |
Female |
16 yr |
(+) |
(±) |
(±) |
(+) |
(n/a) |
(n/a) |
(+) complete data, (±) incomplete data, (n/a) no data
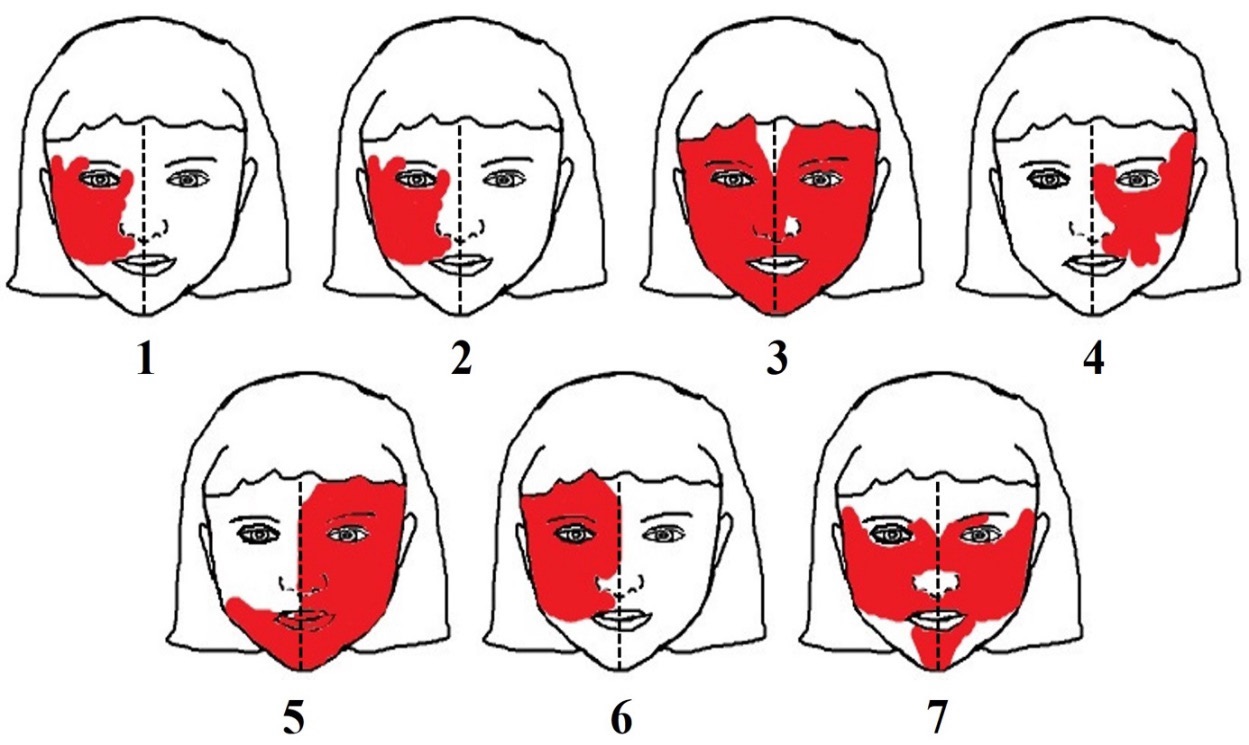
All subjects (100%) had facial PWS distribution pattern at V1 and V2 (Figure 2). The placode distribution patterns showed more specific results, in which the P1b pattern was found in 100% of subjects and P1a pattern in only 57% of subjects. Focal seizure was found in 83% of subjects with rare frequency of seizures. Fine motor disorders occurred in 50% of subjects. Glaucoma occurred in all subjects accompanied by partial or total visual impairment, blindness and/or hemianopsia as ophthalmological deficits (Table 3).
Table 3: Facial PWS characteristics, neurological deficiency, and ophthalmological deficiency.
Facial PWS |
Neurological Deficiency |
Ophthalmological Deficiency |
Distribution pattern (n=7)
V1 7 (100%)
P1a 4 (57%) Facial PWS Score (n=7) |
Type of seizure (n=6)
Seizure frequency (n=4)
Motoric disorder (n=4) |
Intraocular pressure (n=7)
Visus (n=6)
Visual field (n=4) |
There were two subjects with complete data of neurological and ophthalmological deficits, EEG and MRI. In both subjects neurological and MRI scores were assessed. The complete characteristics, scores, and diagnostic examination results of these subjects are presented in Table 4. Epileptiform finding in EEG examination was identified in three subjects and only one subject showed normal EEG results. Other diagnostic examination data collected included IQ test, from which only 1 subject had the result of 50 IQ score.
Table 4: Characteristic scores and subject investigation scores.
|
Facial PWS Score |
MRI Score |
Neurological Score |
Epileptiform Waves |
Cerebral Cortex Involvement |
1 |
18 |
12 |
9 |
(+) |
Frontal, Temporal, Parietal, Occipital |
2 |
8 |
2 |
7 |
(-) |
Occipital |
Discussion
Sturge-Weber syndrome involves neurocutaneous defects caused by GNAQ gene mutations that encode the Gαq protein (alpha subunit of the guanosine-5-triphosphate protein). Stimulation of the G protein coupled receptor (GPCR) causes phosphorylation of guanosine diphosphate (GDP) that binds to the Gαq protein to form guanosine triphosphate (GTP). The GαqGTP component is an active form that activates the mitogen activated protein kinase (MAPK) pathway. The MAPK pathway plays a role in proliferation and vascular growth by regulating transcription factors and preventing apoptosis. Mutations in GNAQ cause hyperproliferation and apoptosis inhibition in vasculogenesis which leads to vascular malformation [2,6].
The GNAQ mutations in the progenitor cell are thought to play a role in the extent of vascular malformations. Earlier mutation will cause vascular malformations in more organs compared to late mutation [2]. Roach et al. classified SWS into 3 types: (1) Type I (Classic) which is the most common type found with vascular malformations in the form of facial PWS, leptomeningeal angioma (brain), and angiomas in choroid (eyes), (2) Type II vascular malformations in the form of facial PWS and choroidal angioma, but without leptomeningeal angioma, and (3) Type III (forme fruste) is the rarest type with vascular malformation in the form of leptomeningeal angioma only. The hypothesis about the timing of this mutation can explain all three types of SWS with the involvement of these varied organs [7].
Our study found that the distribution patterns of trigeminal and placode were almost similar. However, the placode distribution pattern showed more specific results, in which 7 subjects with V1 facial PWS can be specified into 7 subjects with P1b facial PWS only and 4 subjects with P1a and P1b. This result is in line with previous studies [3], since embryologically the facial skin area of P1a and P1b vascularization are derived from the same neural crest as the cerebral cortex and eye. This similarity in vascularization causes the placode pattern to be more specific than the trigeminal pattern [3]. The involvement of facial PWS in V1 , P1a and P1b patterns as SWS's "high risk" areas were also found in all subjects, similar with the cases described by Dymerska et al [4].
Vascular malformation in the form of leptomeningeal angiomatosis in SWS is a major cause of neurological deficits. Parsa proposed the absence of any bridging vein as a cause of leptomeningeal angiomatosis in SWS [8]. If venous blood flow from the cortical vein to the superior sagittal sinus is impaired due to the absence of a bridging vein, this condition results in enlargement of other leptomeningeal blood vessels as compensation. This compensatory mechanism leads to increased venous pressure accompanied by decreased arterial perfusion and hypoxic ischemia in the involved brain lobe. In the acute phase, symptoms of seizures can arise which will further increase the oxygen demand and the brain’s metabolic rate. In the late phase, as the seizures occur repeatedly and the brain cannot compensate, progressive atrophy and calcification in the cerebral cortex will occur ranging from acute stroke-like episodes to motor disorders manifestations [4,8].
Neurological deficits correlate with the location and extent of brain involvement. Generalized seizures with frequent, uncontrolled frequency and accompanied by motor disorders indicate extensive and progressive brain involvement [9]. The neurological deficits found in our study included focal seizures in five patients (83%). Fine motor disorders are the most severe degree found in motoric function deficits. Limited and non-progressive brain involvement was found in the majority of subjects. Most of the subjects aged more than 6 years old had neurological deficits that were non-progressive similar with the study of Kavanaugh et al [5]. Our study found that the progression of neurological deficits may be continued in subjects aged less than 6 years old with frequent generalized type seizures.
The choroid layer is a vascular-rich layer between the sclera and retina of the eye. Vascular malformations in the form of angiomatosis of the choroid will cause mechanical angular obstruction in the eye and increasing episclere venous pressure that can manifest clinically as visual field defects and glaucoma. Retinal detachment may also occur due to untreated progressive glaucoma and lead to blindness [10,11]. Glaucoma was found in all of our subjects as the main ophthalmological deficit. Decreased visual and partial hemianopsia were found as the mildest degree of glaucoma progression. These results are similar with the study conducted by Bachur et al., in which glaucoma in the early phase of SWS was asymptomatic. Patients realized the presence of an ophthalmological deficit only when the disease had progressed and this delay made the ophthalmologic examination performed late [9]. Diagnostic and therapeutic intervention delays can result in total blindness and hemianopsia as seen in our patients.
Facial PWS scores along with MRI scores may predict neurological and ophthalmological deficits that are described by neurological scores [4]. Scoring assessments in two of our patients showed that PWS and MRI scores have a positive correlation with neurological scores, similar with the study conducted by Bachur et al [9]. Higher neurological scores were also found in subjects with more extensive lesions in the cerebral cortex [9]. None of subjects have unique facial PWS distribution patterns limited to the P1a distribution, which tend to have very low facial PWS scores with very high neurological scores [4].
EEG abnormalities in SWS were found in the form of asymmetric epileptiform waves ipsilateral with facial PWS. This epileptiform finding becoming more abnormal with more frequent epileptiform activity, resembles the extent of brain involvement that progressively increases with age [12,13]. Our study found that epileptiform findings were in line with the extent of brain involvement, which in one subject with all four brain regions involvement showed EEG abnormalities. No EEG abnormality was found in subjects with only involvement of the occipital region. Neurological and ophthalmological deficits with low intelligence (IQ 50) were also found in one patient with no EEG abnormalities. Previous study reported that white matter focal damage in just one brain region was enough to reduce IQ [5].
Some of the limitations of our study were incomplete data from neurological, ophthalmological, and radiological examinations. The limitations of MRI data also made all subjects to be diagnosed as type I SWS (classic) and there was a possibility that non-classical SWS (types II and III) could be missed. MRI examinations for neurological and ophthalmological deficits are recommended to be performed to provide a better clinical assessment, prognosis, and management of SWS cases. Our study demonstrated that the characteristics of facial PWS, neurological, and ophthalmological deficits are clinical features that were associated with the pathogenesis and severity of SWS. Facial PWS is the most visible sign in patients with SWS and can be an indicator for screening of neurological and ophthalmological deficits. Examinations of EEG and MRI play an important role in screening of brain function deficits and assessing structural abnormalities in SWS.
Conclusion
Sturge-Weber syndrome involves neurocutaneous defects characterized by facial capillary malformations in the form of a PWS accompanied with vascular malformations in the brain and ipsilateral eye. The facial PWS is one of the SWS components which can be an initial marker and help to predict the severity of SWS. Pathogenesis and degree of severity in SWS were related to facial PWS, neurological and ophthalmological deficits. Therefore, facial PWS can be the initial marker and help predict the severity of SWS.
Acknowledgements: The authors want to thank the staff at Klinik Bahasa, Office of Research and Publication, FKKMK-UGM who kindly provided proofreading assistance.
References
- Comi AM. Update on Sturge-Weber syndrome: Diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007; 5: 257-64.
- Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 2013; 368: 1971-9.
- Waelchii R, Aylett SE, Robinson K, Chong WK, Martinez AE, Kinsler VA. New vascular classification of port-wine stains: Improving prediction of Sturge-Weber risk. Br J Dermatol. 2014; 171: 861-7.
- Dymerska M, Kirkorian AY, Offermann EA, Lin DD, Comi AM, Cohen BA. Size of facial port-wine birthmark may predict neurologic outcome in Sturge-Weber syndrome. J Pediatr. 2017; 188: 205-9.
- Kavanaugh B, Sreenivasan A, Bachur C, Papazoglou A, Comi A, Zabel TA. Intellectual and adaptive functioning in Sturge-Weber Syndrome. Child Neuropsychol. 2016; 22: 635-48.
- Nguyen V, Hochman M, Mihm MC, Jr., Nelson JS, Tan W. The pathogenesis of port wine stain and Sturge Weber syndrome: Complex interactions between genetic alterations and aberrant MAPK and PI3K activation. Int J Mol Sci. 2019; 20: 2243-60.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992; 39: 591-620.
- Parsa CF. Focal venous hypertension as a pathophysiologic mechanism for tissue hypertrophy, port-wine stains, the SturgeWeber syndrome, and related disorders: Proof of concept with novel hypothesis for underlying etiological cause. Trans Am Ophthalmol Soc 2013; 111: 180-215.
- Bachur CD, Comi AM. Sturge-weber syndrome. Curr Treat Options Neurol 2013; 15: 607-17.
- Comi A. Current therapeutic options in Sturge-Weber syndrome. Semin Pediatr Neurol 2015; 22: 295-301.
- Sharan S, Swamy B, Taranath DA, Jamieson R, Yu T, Wargon O, et al. Port-wine vascular malformations and glaucoma risk in Sturge-Weber syndrome. J AAPOS. 2009; 13: 374-8.
- Jansen FE, van Huffelen AC, Witkamp T, Couperus A, Teunissen N, Wieneke GH, et al. Diazepam-enhanced beta activity in Sturge Weber syndrome: Its diagnostic significance in comparison with MRI. Clin Neurophysiol. 2002; 113: 1025-9.
- Zallmann M, Leventer RJ, Mackay MT, Ditchfield M, Bekhor PS, Su JC. Screening for Sturge-Weber syndrome: A state-of-the-art review. Pediatr Dermatol. 2018; 35: 30-42.