
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Kindler syndrome: A rare case report
Apoorva D Chopkar1; Sakshi S Malpani1; Bhagyashree B Supekar2*; Jayesh I Mukhi3
1 Junior Resident, Department of Dermatology, Venereology, Leprosy, Government Medical College and Hospital, Nagpur, Maharashtra, India.
2 Assistant Professor, Department of Dermatology, Venereology, Leprosy, Government Medical College and Hospital, Nagpur, Maharashtra, India.
3 Professor and Head of Department, Department of Dermatology, Venereology, Leprosy, Government Medical College and Hospital, Nagpur, Maharashtra, India.
*Corresponding Author: Bhagyashree B Supekar
Assistant Professor, Department of Dermatology,
Venereology, Leprosy, Government Medical College and
Hospital, Nagpur, Maharashtra, India.
Email: bhagyashreesupekar.23@gmail.com
Received : Aug 20, 2021
Accepted : Sep 28, 2021
Published : Oct 05, 2021
Archived : www.jcimcr.org
Copyright : © Supekar BB (2021).
Abstract
Kindler Syndrome (KS) is a rare hereditary disorder characterized by acral blistering of infancy and childhood, photosensitivity, progressive poikiloderma, and cutaneous atrophy. We report this case of KS in a 4 year old female child on account of its rarity
Keywords: Kindler syndrome; acral blistering; pokiloderma; photosensitivity
Citation: Chopkar AD, Malpani SS, Supekar BB, Mukhi JI. Kindler syndrome: A rare case report. J Clin Images Med Case Rep. 2021; 2(5): 1344.
Introduction
KS is an autosomal recessive genodermatosis, a subtype of Epidermolysisbullosa (EB). While the levels of cleft in simplex, junctional and dystrophic types of EBare established, KS may have multiple cleft levels. Clinically, the child presents with acral blistering of infancy and childhood, photosensitivity, progressive poikiloderma, and cutaneous atrophy. Less than 250 cases have been reported in literature since Theresa Kindler’s original description 1954.
Case presentation
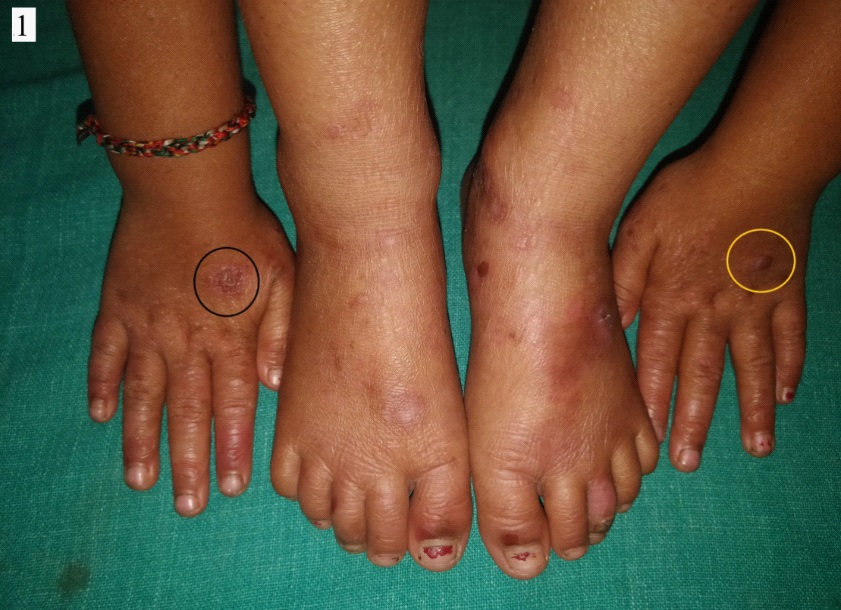
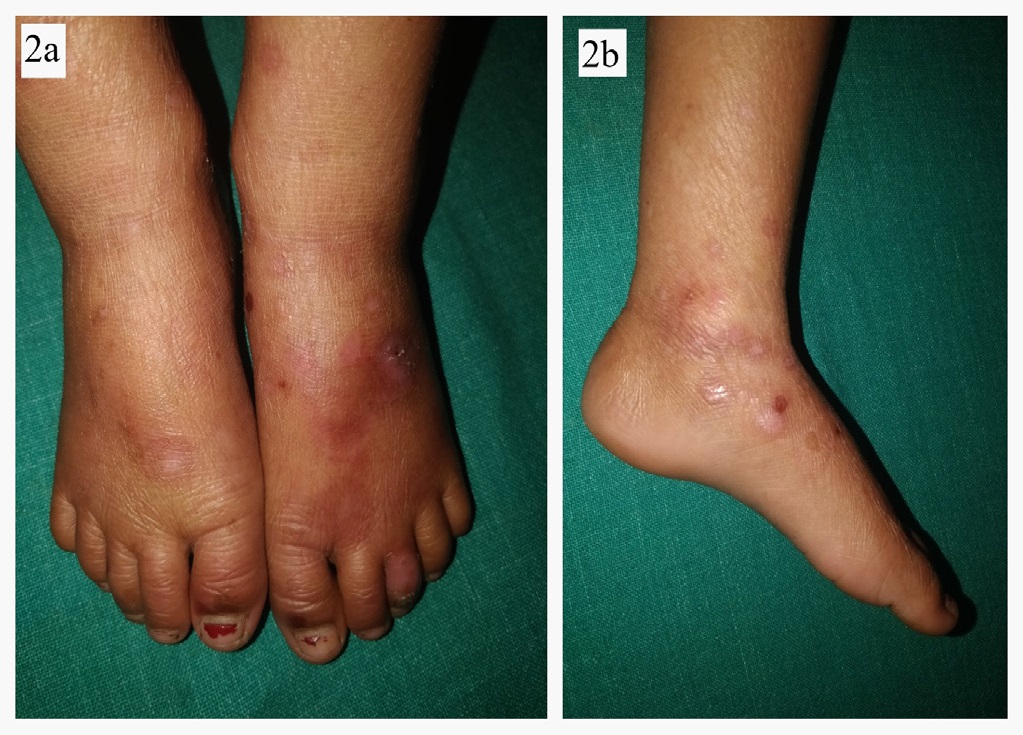
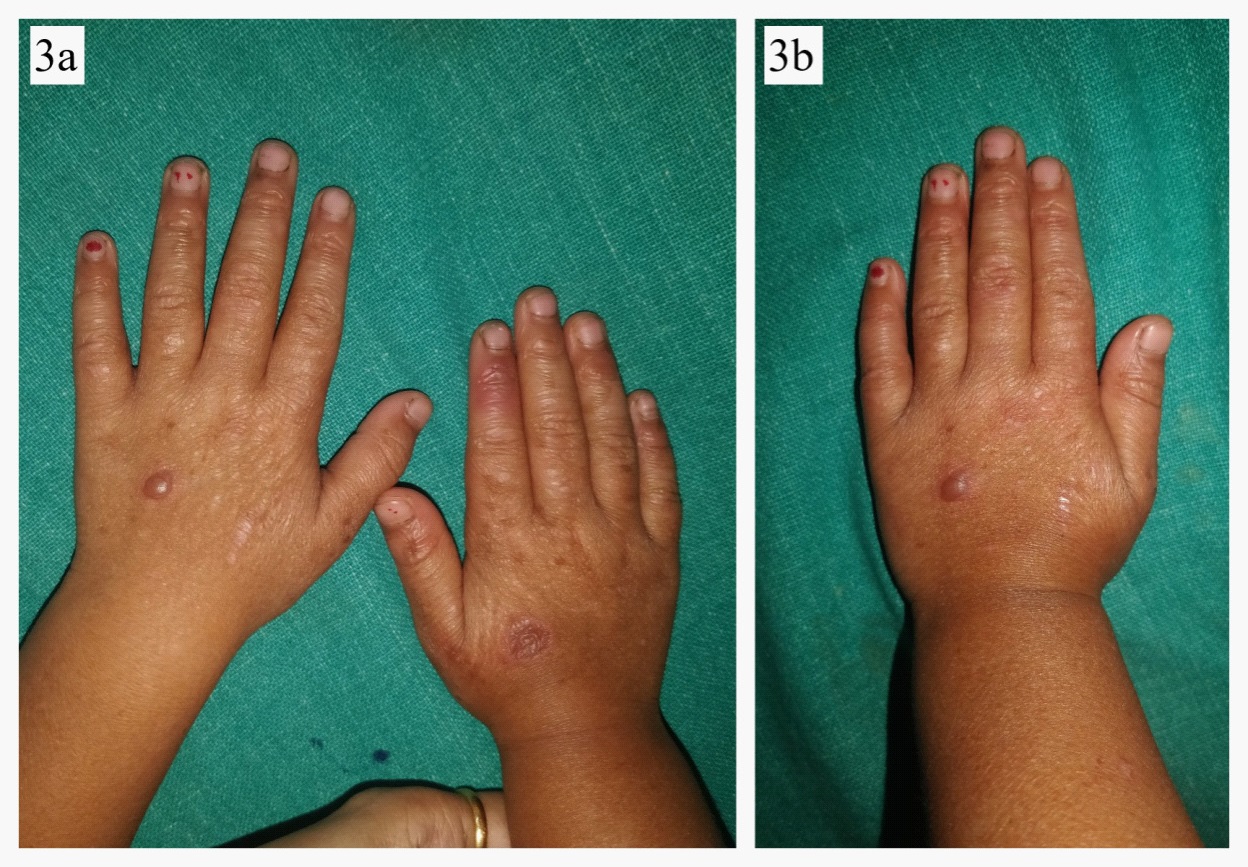
A 4 year old female child born out of a non-consanguineous marriage was brought by her parents with history of fluid filled lesions over hands and feet, followed by discoloration and thin skin over hands and feet since birth. On enquiry, she had history of multiple episodes of these fluid filled lesions without any antecedent trauma, and this tendency to form blistering had reduced with age. She also complained of redness and burning sensation over exposed areas on sun exposure. There was no history of difficulty in deglutition, micturation, or vision. There was no history of similar complaints in family members. On cutaneous examination, there were few intact vesicles on the dorsum of hands and feet overlying severely atrophic cigarette paper like skin (Figures 1,2a,b,3a,b). Diffuse poikiloderma predominantly affecting face was noted (Figure 4a,b). Scalp hair were normal in colour, thickness and growth pattern. On oral examination, calculi and mild gingival inflammation on upper and lower anterior region were present. Nails were normal. Ocular and external genital examination and rest cutaneous examination were unremarkable.
On the basis of history and clinical examination, differential diagnoses of KS, EB with mottled pigmentation and porphyriacutaneatarda were considered.
Complete blood counts and urine analysis were normal. Radiological screening was unremarkable. Urine porphyrin levels were within normal limits. Parents of the child did not consent for skin biopsy, Transmission Electron Microscopy (TEM) and genetic analysis. Patient was treated symptomatically. She was prescribed a broad spectrum sunscreen, topical antibiotics and emollients and was explained about trauma avoidance and blister care. Unfortunately, she was lost to follow up due to the ongoing COVID-19 pandemic.
Discussion
KS was first described by Theresa Kindler in 1954, presenting as simultaneous occurrence of poikiloderma congenitale and hereditary epidermolysis bullosa [1]. In 1971, Weary P, et al reported a widespread eczematous and acral blistering disorders of infancy and childhood with subsequent development of poikiloderma and various acralkeratoses, which he called hereditary acrokeratotic poikiloderma [2]. It had been a matter of widespread debate whether these two entities were separate or belonged to the same spectrum, until recently, when the genetic basis and molecular mechanisms underlying KS were discovered. In 2003, the genetic defect of the syndrome was first localised to loss-of-function mutations in the gene KIND1 mapped to chromosome 20p12.3 [3]. It codes for kindlin-1 protein, expressed particularlyin the basal keratinocytes, loss of which results in defects in actin-extracellular matrix linkages and abnormal skin fragility [4].The largest cluster of KS has been reported from the Ngobe Bugle tribe in the Bocas Del Toro province of Panama, comprising of 26 cases [5].
The typical clinical findings of KS include:
- Acral blistering tendency is usually the earliest feature,
manifesting at birth or early days of life, which subsides
as child grows up. It occurs unprovoked or secondary to
trivial trauma.
- Severe photosensitivity in response to minimal sun exposure.
- Diffuse skin atrophy with cigarette paper consistency, typically pronounced over the dorsum of hands, and feet and
abdomen, mostly occurring during adolescence but have
also been observed in infants as young as 3 months.
- Progressive pokiloderma, prominent over lateral side of
face and neck.
All the 4 above mentioned findings were present in our case. Apart from these, KS may also present with variable mucosal involvement, ranging from xerostomia, angular cheilitis to laryngeal, esophageal, and anal swelling and stenosis [6]. Dental caries, periodontal disease, glandular cheilitis, and leukokeratosis of lips could be present upon oral examination [7].Our case had gingival inflammation and calculi. Syndactyly, sclerotic wrist bands, nail dystrophy, palmoplantar keratoderma, pseudoainhum may be present [8].On ocular examination, corneal opacities, ectropion, thickened corneal nerves may be present [9]. Examination of external genitalia may show phimosis in males and labial agglutination in females [10]. Skeletal examination may reveal a dome shaped skull and bifid ribs [9].
Angelova-Fischer, et al devised a diagnostic criteria in 2005 which has been addressed in Table 1 [11]. As can be surmised from the table, our case fulfils all 5 major criteria, making the diagnosis of KS certain. Diagnosis is mainly clinical and differential diagnoses of KS have been summarized in Table 2. Light microscopic findings include epidermal atrophy, vacuolar alteration with cleft formation, and colloid bodies. TEM of vesicular specimen will show various planes of cleavage at the basement membrane zone with reduplication of lamina densa. Immunostaining with anti-kindlin-1 antibody shows decreased epidermal staining. Skin ultrastructure study shows cleavage planes in variable locations, mostly localized to the lowermost portion of the basal keratinocyte layer. This phenotype correlates with genotype as the mutation results in structural defects in anchoring of actin filaments in the lowermost portion of keratinocytes [12].
Table 1:
Major findings |
Minor findings |
Associated findings |
Acral blistering in infancy and childhood |
Syndactyly |
Nail dystrophy |
Progressive poikiloderma |
Mucosal involvement |
Ectropion of lower lid |
Skin atrophy |
|
Palmoplantarkeratoderma |
Abnormal photosensitivity |
|
Pseudoainhum |
Gingival fragility and/or swelling |
|
Leukokeratosis of lips |
|
|
Squamous cell carcinoma |
|
|
Anhidrosis/hypohidrosis |
|
|
Skeletal abnormalities |
|
|
Poor dentition/dental caries/periodontosis |
CRITERIA: Certain= 4 major |
||
Table 2:
Disorder |
Acral blistering |
Photosensitivity |
Poikiloderma |
Skin atrophy |
Comments |
Other forms of Epidermolysisbullosa (EB) |
Present |
Absent |
Present |
Present |
Blistering will be trauma induced and persistant. Findings will differ according to type of EB- simplex, dystrophic, junctional; localized/generalized involvement; systemic involvement |
Porphyria cutanea tarda |
Present |
Present |
Absent |
Present |
Pseudosclerodermatous scarring, polycythemic facial plethora, non virilising hypertrichosis |
Weary Kindler syndrome |
Absent |
Absent |
Present (flexural) |
Present |
Sclerotic bands, poor dentition, and, occasionally, calcinosis |
Xeroderma |
Absent |
Present |
Present |
Present |
early-onset skin cancers and multiple neurologic abnormalities |
Bloom syndrome |
Absent |
Present |
Absent |
Absent |
Telangectasias, Short stature, recurrent infections, and increased frequency of hematological malignancies |
Rothmund Thompson syndrome |
|
Present |
Present |
|
Short stature, sparse scalp hair, sparse or reduced eyelashes and eyebrows, hypogonadism, premature aging, and juvenile cataracts |
Dyskeratosis |
Absent |
Absent |
Absent |
Absent |
Reticulated hyperpigmentation (not true poikiloderma), nail dystrophy, and leukoplakia |
Treatment of KS is mainly symptomatic. Medical care includes prevention of skin trauma which will bring down new blister formation, proper wound care and dressings to hasten healing and prevent of infections, use of appropriate sunscreens, good nutrition, surveillance for extracutaneous complications, psychosocial support and genetic counselling. Antioxidants and flavinoidluteolin which reduce UV-B induced keratinocyte apoptosis may be upcoming therapies [13]. Telangiectasias can be treated with vascular lasers. Surgical correction of urethral, anal, or esophageal stenosis may be done as and when required.
The treatment is multidisciplinary and consultations with various departments, primarily paediatrics, dentistry, ophthalmology, gastroenterology, urology, gynecology, and psychiatry should be made when needed.
Conclusion
We report this case because it’s rare and also needs to be differentiated from a plethora of similar disorders. Due to its wide spectrum of features, KS is associated with significant morbidity and mortality. As there is no definitive treatment, management is mainly directed towards reducing morbidity and improving the quality of life.
References
- Kindler T. Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy. Br J Dermatol. 1954; 66: 104–11.
- Weary P, Manley WJr, Graham G. Hereditary acrokeratoticpoikiloderma. Arch Dermatol. 1971; 103: 409–22.
- Siegel DH, Ashton GH, Penagos HG, Lee JV, Feiler HS, Wilhelmsen KC, et al. Loss of kindlin-1, a human homolog of the Caenorrhabditiselegansactin-extracellularmatrix linker protein UNC112, causes Kindler syndrome. Am J Human Genet. 2003; 73: 174-87.
- Martignago BC, Lai-Cheong JE, Liu L, McGrath JA, Cestari TF. Recurrent KINDIN1 (C20orf42) gene mutation, c.676insC, in Brazilian pedigree with Kindler syndrome. Br J Dermatol. 2007; 157: 1281-4.
- Penagos H, Jaen M, Sancho M et al. Kindler syndrome in native Americans from Panama. Arch Dermatol 2004; 140: 939–44.
- Ohashi A, Kiniwa Y, Okuyama R, Kosho T, Suga T, Has C, et al. A case of Kindler syndrome with severe esophagealstenosis. Int J Dermatol. 2015; 54: e106-8.
- Barbosa NM, Visioli F, Martins MD, Martins MA, Munerato MC. Oral manifestations in Kindler syndrome: case report and discussion of literature findings. Spec Care Dentist. 2016; 36: 223-30.
- Al Aboud K, Al Hawsawi K, Al Aboud D, Al Githami A. Kindler syndrome in a Saudi kindred. Clin Exp Dermatol. 2002; 27: 673–6.
- Sharma R, Mahajan V, Sharma N, Sharma A. Kindler syndrome. Int J Dermatol. 2003; 42: 727–32.
- Krishna CV, Parmar NV, Has C. Kindler syndrome with severe mucosal involvement in childhood. Clin Exp Dermatol. 2014; 39: 340-3.
- I. Angelova-Fischer, J. Kazandjieva, S. Vassileva, A. Dourmishev. Kindler syndrome: a case report and proposal for clinical diagnostic criteria. ActaDermatoven APA. 2005; 14: 2.
- Burch JM, Fassihi H, Jones CA, Mengshol SC, Fitzpatrick JE, et al. Kindler syndrome: A new mutation and new diagnostic possibilities. Arch Dermatol. 2006; 142: 620-4.
- Maier K, He Y, Wolfe U, Esser PR, Brummer T, Schempp C, et al. UVB induced cutaneous inflammation and prospects for antioxidant treatment in Kindler syndrome. Hum Mol Genet. 2016.