
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Suprarenal unicentric Castelman’s disease: A rare case report
Mohammed Arafath Ali; Ramachandra C; Uday Karjol; Ravi Arjunan; Syed Altaf; Srinivas C; Akkamahadevi S Patil; Pavan Sugoor*
Kidwai Memorial Institute of Oncology, Department of Surgical Oncology, Bengaluru, Karnataka, India.
*Corresponding Author: Pavan Sugoor
Kidwai Memorial Institute of Oncology, Department
of Surgical Oncology, Bengaluru, Karnataka, India.
Email: pavansugoor26@gmail.com
Received : Aug 30, 2021
Accepted : Oct 04, 2021
Published : Oct 11, 2021
Archived : www.jcimcr.org
Copyright : © Sugoor P (2021).
Abstract
Castleman Disease (CD) is a rare non-clonal lymphoproliferative disorder. The clinical presentation of CD often overlaps with autoimmune, infectious, or other malignant diseases. The diagnosis is confirmed by a biopsy of the affected lymph-node tissue. A 39-year-old gentleman was evaluated for intractable abdominal pain fatigue of 2 months duration. CT scan showed a 4 X 4 X 5 cm right suprarenal mass. Laparotomy and near total excision was performed which showed reactive B cells expressing CD20 and PAX5, favoring CD. Patient received adjuvant 6 cycles of Rituximab and steroids. A follow up CT scan after 3 months showed a near complete resolution.
We herein report a rare case of retroperitoneal Castleman’s disease.
Keywords: Castelman’s disease; unicentric; multicentric; lymphoproliferative disease; rituximab.
Citation: Ali MA, Ramachandra C, Karjol U, Arjunan R, Altaf S. Suprarenal unicentric Castelman’s disease: A rare case report. J Clin Images Med Case Rep. 2021; 2(5): 1353.
Introduction
Castelman’s disease (CD) was first documented and described by Dr. Benjamin Castelman as a rare lymphoproliferative disorder [1]. CD is a heterogeneous group of disorders and can present as single enlarged lymph node i.e. Unicenteric or as a Multicenteric lymphadenopathy affecting multiple lymph node stations.
Multicenteric disease is usually associated with constitutional symptoms like fatigue, loss of weight, pyrexia, cytopenias and multiple organ dysfunction and is believed to be driven by IL-6 and many other cytokines [2]. However 50% of multicentric disease is caused by uncontrolled Human Herpes Virus – 8 (HHV8) infection in immunocompromised patients whereas HHV-8 negative multicenteric disease is considered idiopathic [3].
Unicenteric disease presents as a solitary enlarged lymph node or as multiple enlarged lymph nodes in a single lymph node station [1]. The clinical course is generally indolent with slow and gradual enlargement of lymph nodes [4]. The average size of lymph node is larger in unicenteric disease than multicenteric disease [5].
Case presentation
A 39 year old gentleman presented to us with complaints of intractable abdominal pain, fatigue, lethargy and anorexia for 2 months. The patient had no comorbidities. Physical examination was unremarkable and on abdominal examination, mild tenderness was elicited in right hypochondrium. All the blood investigations and markers were in normal range. A CT scan showed a 3 X 4.2 X 5.2 cm homogenously enhancing soft tissue mass in the right suprarenal location, in close relation to inferior vena cava (Figure 1-3).
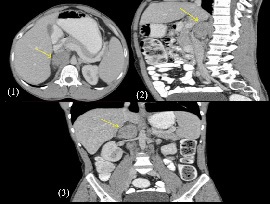
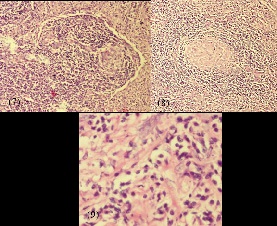
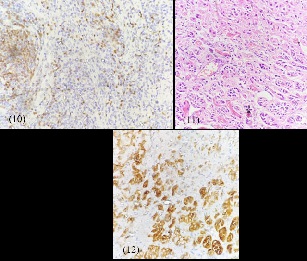
Laparotomy and near total excision of the lymph nodal mass was performed as image guided biopsy was technically challenging. Histopathological examination demonstrated lymph nodes with follicular hyperplasia admixed with numerous plasma cells and eosinophils, ateric lymphoid follicles with hyalinised blood vessels with nests of paraganglions showing the possibility of CD. Immunohistochemistry stains demonstrated reactive B cells expressing CD20 and PAX5 with absence of CD30 and LMPI expression, favouring CD (Figure 7-12).
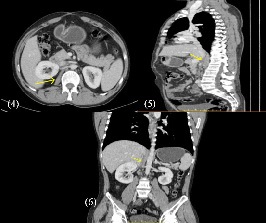
A response assessment CT scan at the end of 6 cycles of rituximab and steroids showed near complete resolution (Figure 4-6) and a good clinical response.
Discussion
Unicenteric disease is usually insidious in onset and involves a single lymph nodal station, it can be diagnosed at any age ranging from 2 – 84 years with a median age at diagnosis of 34 years. Unicenteric disease has a slight female preponderance.
There are no known epidemiologic factors for the development of unicenteric disease and are almost always HHV – 8 negative [6].
Due to the indolent course of unicenteric disease its diagnosis is usually incidental as lymphadenopathy is often asymptomatic in nature, however the symptoms can arise if there is mass effect on adjacent structures from enlarged nodes for example on airways, ureters or adjacent neurovascular bundle.
Most common sites of involvement of unicenteric CD are mediastinum (29%), neck (23%), abdomen (21%) and retroperitonium (17%) and other uncommon sites like axilla, inguinal region, orbits, nasopharynx and small intestine [7].
TURBT encompasses the risk of stimulation of the obturator nerve, which is a significant concern during surgery. It can lead to complications like bladder wall perforation and vascular injury, so EA combined with ONB is safely applied to prevent the obturator jerking. It precludes adductor spasms.
Unicenteric disease is divided into 3 subtypes based on histopathological features (i) Hyaline vascular subtype. (ii) Plasmacytic subtype. (iii) Mixed subtype which shows features of both. The hyaline vascular subtype is the most common subtype and is usually not associated with systemic symptoms.
However the patients with plasmacytic or mixed variant of unicenteric CD may have iMCD like constitutional symptoms and deranged laboratory parameters like anemia, elevated ESR, or C reactive protein, hypoalbuminemia [8].
Management of unicenteric disease is based on consideration of the following factors: (I) location of the enlarged lymph node, (II) resectability status of the involved lymph node or group of lymph nodes, (III) degree of compression of neighboring structures or systemic inflammatory syndrome.
Based on the above considerations the management differs.
(i) Resectable UCD: The recommended first line treatment is
surgical extirpation which eliminates any systemic symptomatology and laboratory abnormalities and is found to
have high response rate [9]. Recurrence is uncommon after complete resection [10].
(ii) Unresectable UCD: Generally encountered in the mediastinum adjacent to major blood vessels or bronchi. This
category is further classified into:
(a) Asymptomatic unresectable UCD: As the UCD can remain
stable or slowly grow over a long period of time it is best
to wait and watch [11].
(b) Symptomatic unresectable UCD: Usually due to compression of adjacent vital structures and can be treated with
partial resection in order to reduce the bulk of the disease
without complete removal, it is associated with good results provided the patient is followed up to monitor the
growth [10].
(c) Inflammation related unresectable UCD: It is driven by
IL-6 and is rare in UCD; it is ideally treated with Silutuximab as first line or Tocilizumab as second line before considering surgery [12].
(d) Persistent symptomatic unresectable UCD: Radiotherapy
can be used as an alternative therapy in this subgroup
with persistent symptoms and lymphadenopathy, which persists despite treatment directed at reliving compression or inflammation related symptoms, another alternative approach is treatment with immunomodulators, immunosuppressant like corticosteroids, cyclosporine and
sirolimus [13].
Follow-up of UCD is done 1 – 3 months after initial treatment and the response to the treatment given must be documented by history. Physical exam, laboratory values and imaging. Later UCD patients are followed annually with CT and laboratory studies. Annual imaging may be discontinued after 5 years.
Conclusion
UCD presents with a diagnostic dilemma as just lymphadenopathy may have a differential diagnosis of lymphoma or testicular tumors in males. Castelmans disease must be viewed as diagnosis of exclusion due to the rarity of the disease.
Complete surgical excision is the preferred intervention of UCD and the recurrence of the disease is rare and in patients with inflammation related symptoms IL-6 inhibitors like Rituximab along with steroids are feasible and effective options, as in our case due to difficulty in procurement of first line drugs.
Declarations
Funding: None
Conflict of interest: No potential conflict of interest relevant to this article
Ethics approval: Appropriate statements included
Consent to participate: All patients had consented for the proposed surgery. This is a retrospective analysis of prospectively maintained database which doesn’t necessitate consenting for analysis
Consent for publications: All authors have consented for publication.
References
- Castleman B, Iverson L, Menendez Vp. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956; 9: 822-830.
- Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: Novel insights into biology, pathogenesis, and therapy. Blood. 2014; 123: 2924-2933.
- Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/ idiopathic multicentric Castleman disease. Blood. 2017; 129: 1646-1657.
- Talat N, Schulte KM. Castleman’s disease: systematic analysis of 416 patients from the literature. Oncologist. 2011; 16: 1316-1324.
- Dong Y, Zhang L, Nong L, et al. Effectiveness of rituximab-containing treatment regimens in idiopathic multicentric Castleman disease. Annals of Hematology. 2018; 97: 1641-1647.
- Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015; 56: 1252-1260.
- van Rhee F, Oksenhendler E, Srkalovic G, et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv. 2020; 4: 6039-6050.
- Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV- and HHV-8-negative Castleman disease. Blood. 2017; 129: 1658-1668.
- Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972; 29: 670-683.
- He X, Wang Q, Wu Y, et al. Comprehensive analysis of 225 Castleman’s diseases in the oral maxillofacial and neck region: A rare disease revisited. Clin Oral Investig. 2018; 22: 1285-1295.
- Soumerai JD, Sohani AR, Abramson JS. Diagnosis and management of Castleman disease. Cancer Control. 2014; 21: 266-278.
- Oksenhendler E, Boutboul D, Fajgenbaum D, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol. 2018; 180: 206-216.
- Bowne WB, Lewis JJ, Filippa DA, et al. The management of unicentric and multicentric Castleman’s disease: A report of 16 cases and a review of the literature. Cancer. 1999; 85: 706-717.