
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Lymphangioleiomyomatosis
Klein Dantis1*; Ranganath TG2
1 Thoracic Surgeon, Assistant Professor, Department of Cardiothoracic Surgery, All India Institute of Medical Sciences, Raipur, India.
2 Assistant Professor, Department of Pulmonary medicine, All India Institute of Medical Sciences, Raipur, India.
*Corresponding Author: Klein Dantis
Thoracic Surgeon, Assistant Professor, Department of
Cardiothoracic Surgery, All India Institute of Medical
Sciences, Raipur, India, 492099.
Email: drkleindantis86@gmail.com
Received : Sep 13, 2021
Accepted : Oct 15, 2021
Published : Oct 22, 2021
Archived : www.jcimcr.org
Copyright : © Dantis K (2021).
Abstract
Lymphangioleiomyomatosis (LAM) is a rare systematic neoplastic disease exclusively seen in middle-aged women with an incidence of 5-9 per million. They can occur sporadically or in association with tuberous sclerosis. Histopathological diagnosis is the gold standard. Median transplant-free survival from the time of diagnosis is 23 years. We here by present premenstrual female with history of recurrent dyspnea with differential diagnosis for various interstitial lung disease diagnosed to have LAM. She was managed with bronchodilator therapy and pulmonary rehabilitation as per European respiratory society guidelines.
Keywords: lymphangioleiomyomatosis; interstitial lung disease; uniportal VATS; histopathology
Citation: Dantis K, Ranganath TG. Lymphangioleiomyomatosis. J Clin Images Med Case Rep. 2021; 2(5): 1378.
Introduction
Lymphangioleiomyomatosis (LAM) is a rare systematic neoplastic disease exclusively seen in middle-aged women with an incidence of 5-9 per million [1]. They can occur sporadically or in association with kidney angiomyolipoma or meningioma [2]. They are asymptomatic in the early phase of the disease or manifest as dyspnea, pneumothorax, and recurrent cough with loss of lung function at an accelerated rate. They can express lymphogenic growth factor, vascular endothelial growth factor D (VEGF-D), matrix metalloproteinases, erythropoietin receptors, and several chemokines with access to lymphatic channels or facilitates metastatic spread [1].
Case report
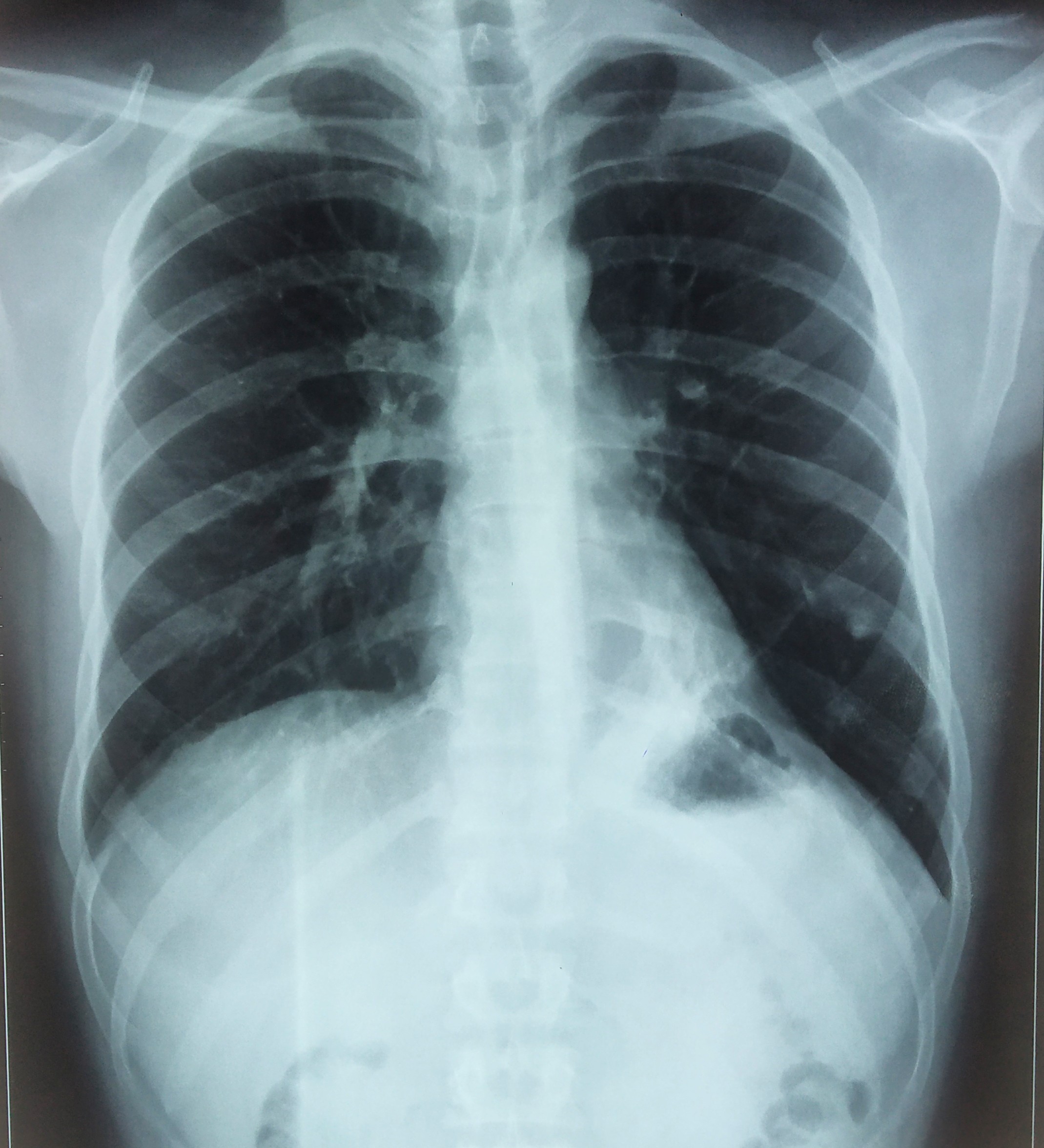
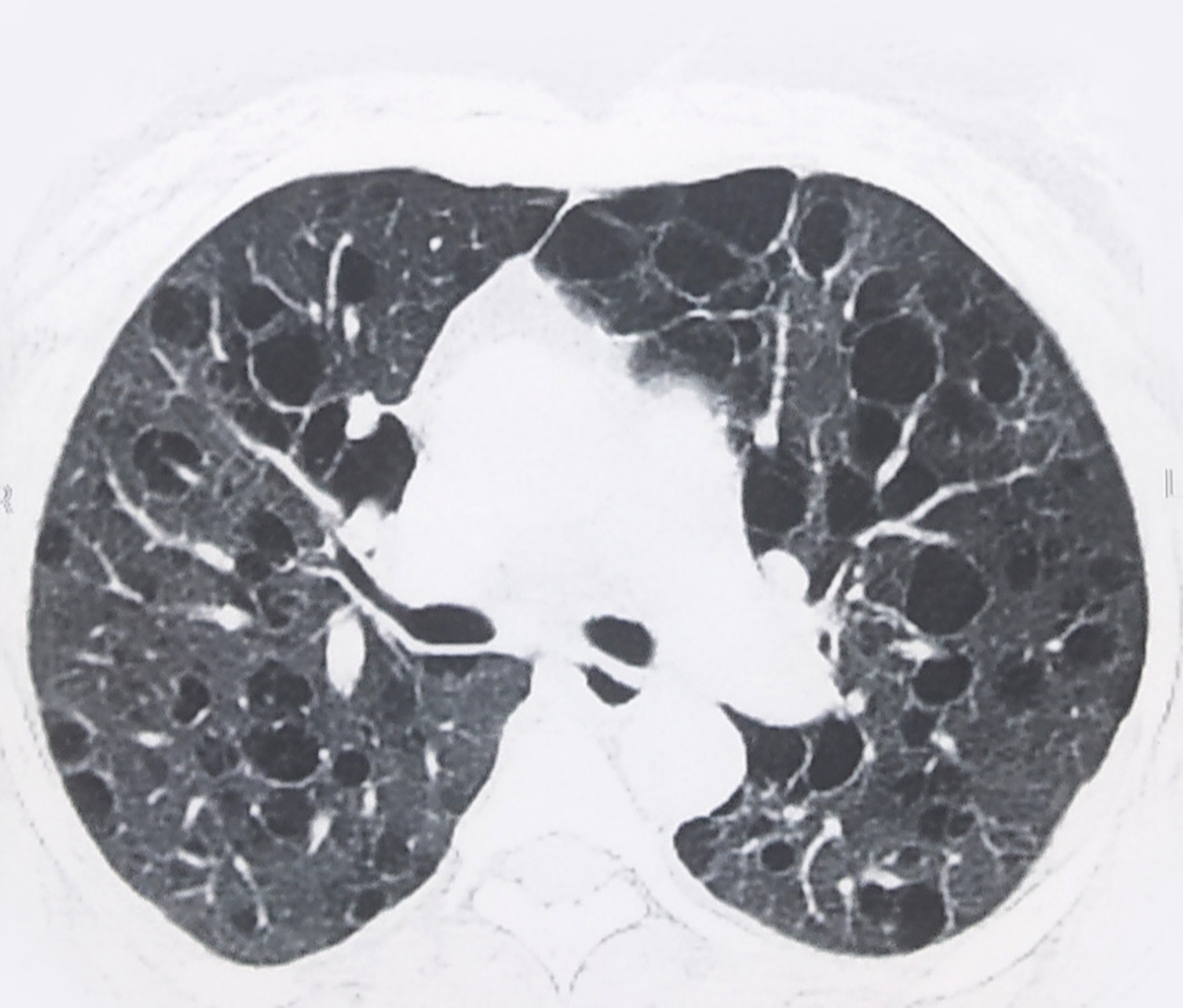
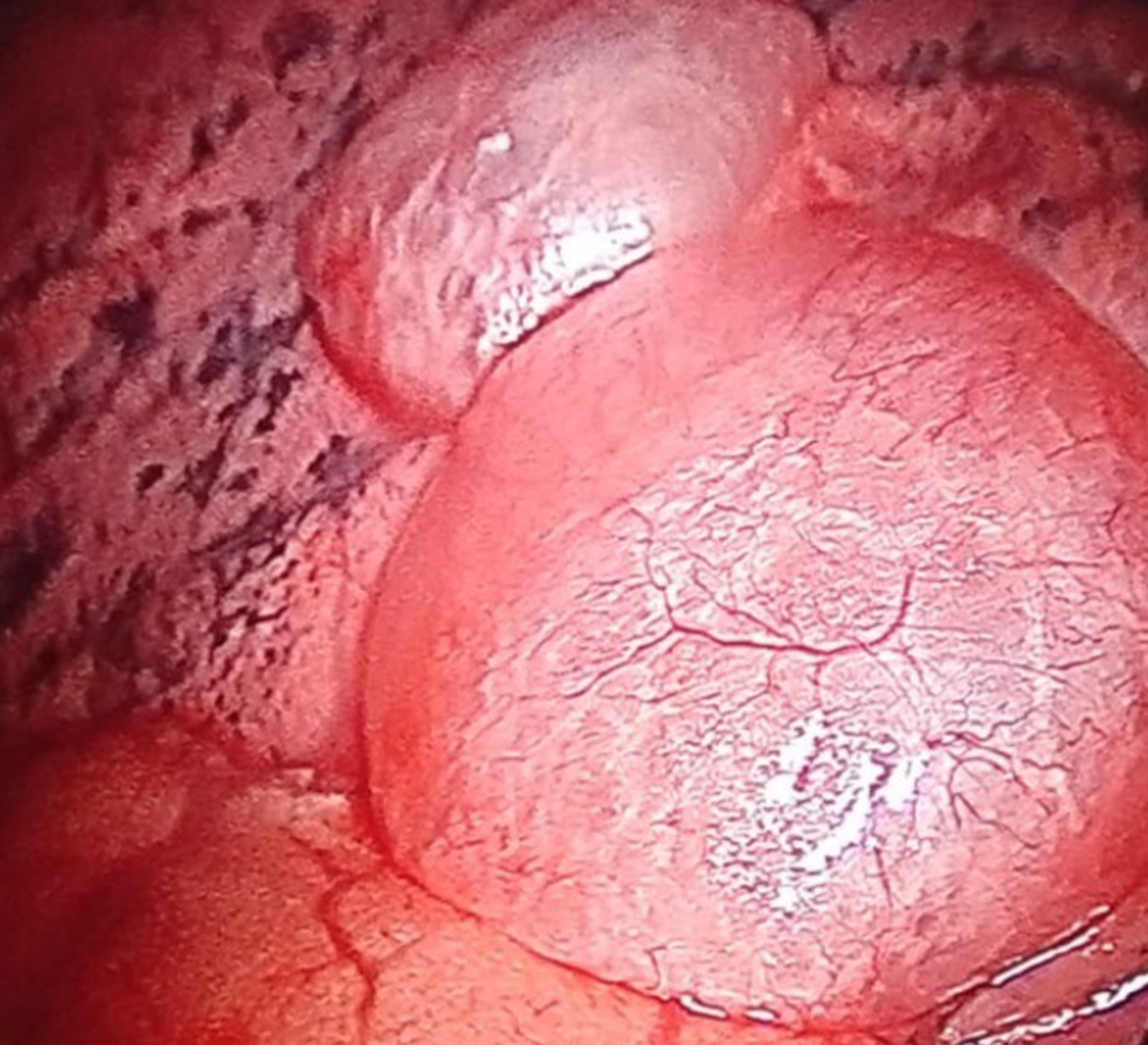
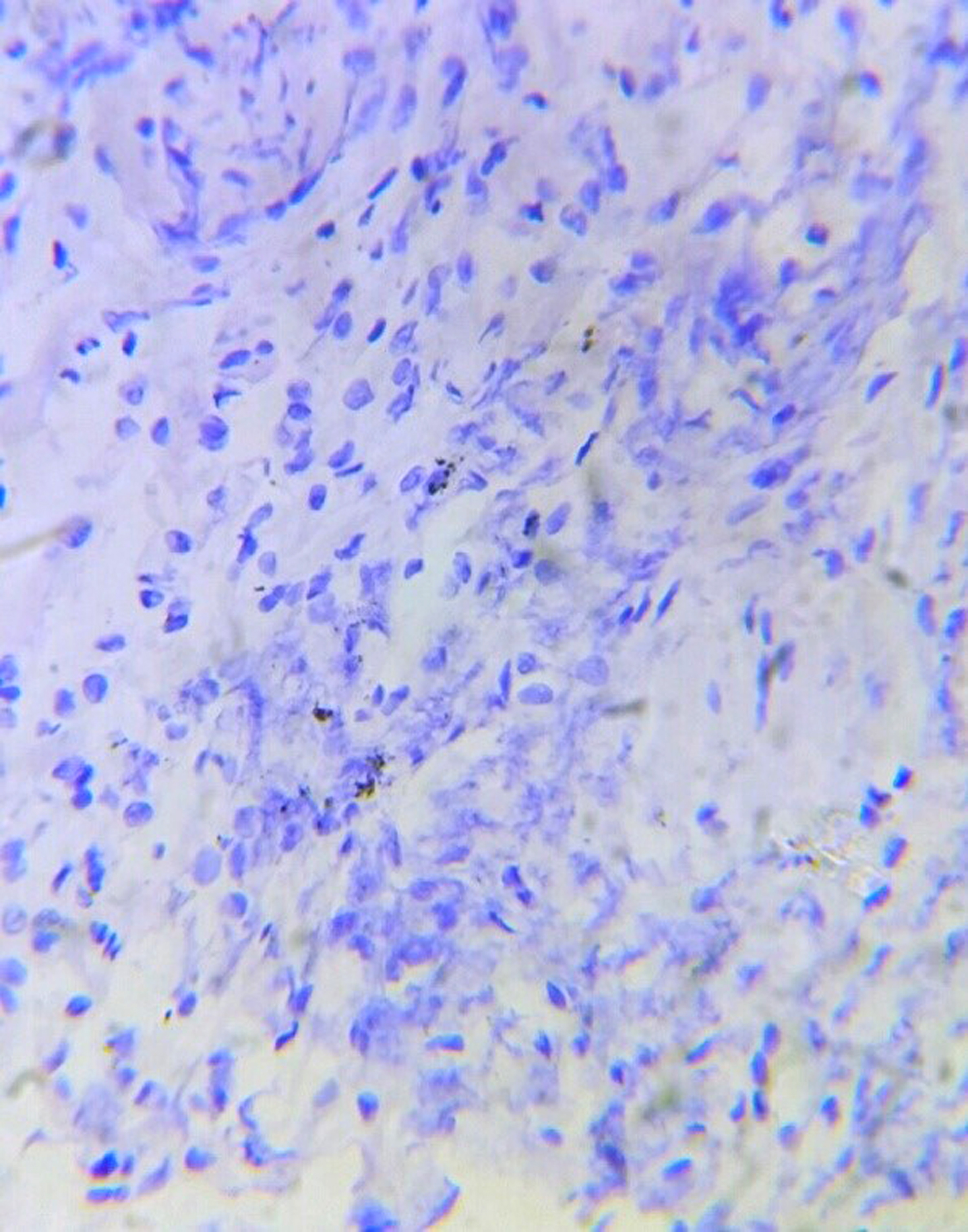
A 46-year old female presented with recurrent dyspnea for two months in the emergency department. Her oxygen saturation was 96% on ambient room air. Her Body Mass Index (BMI) was 25.5 Kg/m2 . She received 2 liters of oxygen/minute with bronchodilator treatment. A chest x-ray showed hazy tiny nodular lesions in the lung parenchyma (Figure 1a). Chest HRCT revealed multiple bilateral cystic lesions throughout the lung parenchyma with the differential diagnosis for interstitial lung diseases - Lymphangioleiomyomatosis (LAM), Langerhans cell histiocytosis (LCH), Lymphoid Interstitial Pneumonia (LIP), and Birt-Hogg-Dube Syndrome (Figure 1b). Her ultrasound abdomen was normal. Pulmonary function test revealed FEV1 - 46% predicted and FVC-60% predicted. Echocardiography did not reveal any abnormality. As the patient had progressive dyspnea, CT-guided needle aspiration was attempted; however termed to be inconclusive hence, she underwent uniportal thoracoscopic wedge biopsy from her right upper and lower lobe under general anesthesia. Intraoperatively, multiple cystic lesions were seen throughout the lung parenchyma (Figure 2a). The postoperative course was uneventful. A single drain placed at the site of the incision was removed on the first postoperative day. Histopathological diagnosis was suggestive of LAM, positive for HMB-45 (Figure 2b). She received bronchodilator treatment for 14 days; however, further bronchodilator treatment or mTOR inhibitors were not considered as she had no severe obstructive symptoms or deteriorating lung function. She received simultaneous pulmonary rehabilitation care and suggested weight loss activity for BMI on overweight classification. Regular follow-up at the 6-month visit was uneventful.
Discussion
LAM management requires an interdisciplinary approach by a pulmonologist, radiologist, oncologist, Thoracic surgeon, and rehabilitation specialist. Multicystic parenchymal disease on high-resolution CT usually points to LAM; however, various differential diagnoses, mainly LCH, LIP, and Birt-Hogg-Dube syndrome, need to be ruled out.
Characteristic CT finding in LAM is multiple small rounded thin-walled cysts measuring 2-10 mm distributed throughout the lung parenchyma equally from central to the periphery with surrounded normal lung parenchyma [1]. In LCH, cysts are small, irregularly marginated, measuring 1-10 mm, confined to the upper lobes sparing costophrenic sinuses with occasionally associated pulmonary trunk enlargement [2]. In LIP, cysts are thin-walled, measuring 30 mm, diffusely distributed, mainly confined to lower lobes along the peribronchovascular bundle, ground-glass opacities and focal consolidation. They are usually associated with connective tissue disorders or immunodeficiency disease [3]. In contrast, in Birt-Hogg-Dube syndrome, cysts are irregular, oversized, unequally distributed with associated renal cell carcinoma or fibrofolliculomas and pathologically confirmed folliculin mutation [4]. Metastatic cystic lesions are seen in 4% of the cases [5]. The most common aetiology is head and neck squamous cell carcinoma detected in 69% of the cases [2].
VEGF-D is a biomarker elevated, especially in LAM, with a 100% specificity, the threshold above 800 pg/ml. However, we did not perform the test as it was expensive from the patient's perspective. A thoracoscopic biopsy followed by histopathological confirmation is the gold standard; however, the patient's tolerability needs to be considered for intubation or non-intubated procedure as transthoracic biopsy provides a yield of 50-60% [6].
As the patient in our case study had recurrent dyspnea with moderate obstruction, as per Global initiative for chronic obstructive lung disease, optimization of pulmonary function was not required; hence, she underwent a thoracoscopic procedure under single lung ventilation. Initial pneumothorax with LAM requires pleurodesis, which was not the presentation; hence, we did not do pleurodesis [7]. In patients with progressive lung function loss, chylous collection, or angiomyolipomas, the Mammalian target rapamycin activation inhibitor (mTOR)-sirolimus and everolimus have a definite role inhibiting the proliferation of LAM cells, which was not required in present case due to moderate obstruction, that is managed with bronchodilators [8]. Median transplant-free survival from the time of diagnosis is 23 years; however, in patients with severe obstructive disease prognosis is morbid [8]. Usually, LAM progresses towards respiratory failure hence pulmonary transplantation is recommended if appropriate, which is not with other cystic diseases with differential diagnosis for LAM [7].
Conclusion
LAM is a rare disease with differential diagnosis for various cystic lung disease. Histopathological diagnosis is must to confirm the diagnosis.
Declarations
Written inform consent was obtained from the patient for the purpose of publication.
Ethical approval is not required from the institution for case report.
Ethical board review is not required from the institution for case report.
Acknowledgements: I would like to thank Melisha R. Pinto (melisha.pinto@gmail.com) for proof reading and editing the manuscript.
Authors contribution: KD drafted the manuscript. Both the authors analysed, interpreted the data.
References
- Xu KF, Feng R, Cui H, Tian X, Wang H, et al. Diffuse cystic lung diseases: Diagnostic Considerations. Semin Respir Crit Care Med. 2016; 37: 457-67.
- Baldi BG, Carvalho CRR, Dias OM, Marchiori E, Hochhegger B. Diffuse cystic lung diseases: differential diagnosis. J Bras Pneumol. 2017; 43: 140-149.
- Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse cystic lung disease. Part II. Am J Respir Crit Care Med. 2015; 192: 17-29.
- Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S, et al., European BHD Consortium. Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol. 2009; 10: 1199-206.
- Seo JB, Im JG, Goo JM, Chung MJ, Kim MY. Atypical pulmonary metastases: spectrum of radiologic findings. Radiographics. 2001; 21: 403-17.
- Johnson SR, Cordier JF, Lazor R, Cottin V, Costabel U, et al. Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010; 35: 14-26.
- Almoosa KF, Ryu JH, Mendez J, Huggins JT, Young LR, Sullivan EJ et al. Management of pneumothorax in lymphangioleiomyomatosis: effects on recurrence and lung transplantation complications.Chest. 129: 1274-81.
- Johnson SR, Taveira-DaSilva AM, Moss J. Lymphangioleiomyomatosis. Clin Chest Med. 2016; 37: 389-403.