
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Paraneoplastic cushing’s syndrome: A diagnostic and therapeutic challenge: A case report and literature review
M Chiboub1*; I Kammoun1; R Gharbi2; H Kammoun2; M Bechikh3; E Haouet3; A Margheli4; H Kandara4
1 Faculty of Medicine, El Manar, Tunis, Tunisia
2 Endocrinology Unit, Department B of Endocrinology, National Institute of Nutrition, Tunis, Tunisia.
3 Department of Pneumology Ibn Nafis, Abderrahman Mami Hospital, Ariana, Tunisia.
4 Department of thoracic surgery, Abderrahman Mami Hospital, Ariana, Tunisia.
*Corresponding Author: Marwa chiboub
Legumes Laboratory, Borj Cedria Biotechnology
Center, University Tunis El Manar, BP 901, 2050,
Hammam Lif, Tunisia.
Email: marwa.chiboub@yahoo.com
Received : Sep 13, 2021
Accepted : Oct 18, 2021
Published : Oct 25, 2021
Archived : www.jcimcr.org
Copyright : © Chiboub M (2021).
Abstract
Ectopic Cushing syndrome (ECS) is difficult to diagnose and has a poor prognosis. We report a case of a 44-year-old male who was admitted for clinical suspicion of Cushing’s syndrome. The investigations confirmed the diagnosis of ECS. No tumor was detected in preliminary imaging tests. Then, the octreoscan showed a fixation in a suspect lung node in the middle lobe. The patient was treated initially with medical drugs, he underwent a right middle lobectomy, and yet histopathology did not reveal any nodule. However, the outcome was the healing of our patient.
We reviewed published articles to access new diagnostic techniques or advantages in the treatment regimen and a prognosis of ECS.
Keywords: ectopic cushing’s syndrome; paraneoplastic syndromes; lung cancer.
Citation: Chiboub M, Kammoun I, Gharbi R, Kammoun H, Bechikh M, et al. Paraneoplastic cushing’s syndrome: A diagnostic and therapeutic challenge: A case report and literature review. J Clin Images Med Case Rep. 2021; 2(5): 1380.
Introduction
Cushing’s syndrome is caused by excess amounts of glucocorticosteroids in the blood, which can be exogenous or endogenous in origin [1]. It may be difficult to miss the diagnosis in its most florid form of major and rapidly evolving hypercorticism. However, the clinical presentation of hypercortisolism associated with this syndrome may be strictly identical to that of Cushing’s disease, hence making diagnosis difficult [2].
Increased endogenous glucocorticosteroids can be either from the pituitary glands (the most common cause), adrenal glands or ectopic secretion of ACTH or CRH, which eventually leads to elevated blood cortisol. According to a recent manuscript, with data from the large ERCUSYN cohort, only 6% of Cushing’s syndrome was caused by ectopic ACTH secretion, whereas 67% had a pituitary origin and 25% adrenal origin [3]. This ectopic secretion is secondary to neuroendocrine tumors of varying size and localization. In adults, the most common tumors causing ectopic ACTH Syndrome are bronchial neuroendocrine tumors, carcinoid of thymus, pancreatic carcinoma and neural tumors.
This report describes a 44-year-old man who presented with clinical signs of hypercorticism and was diagnosed with paraneoplastic Cushing syndrome due to ectopic ACTH production. Highlighting how difficult it might be to determine the source of ACTH secretion and how to manage such a case. The patient approved all medical and surgical procedures and was adherent. The patient’s consent was obtained before writing the case.
Case presentation
A 44-year-old man had a history of 30 pack-years smoking, 11 years of diabetes and a recent high blood pressure poorly controlled with more than 3 antihypertensive drugs. His diabetes was complicated with a bilateral proliferative diabetic retinopathy, diabetic nephropathy with a severe nephrotic syndrome without renal failure. He was admitted in the internal medicine department for an amyotrophy of the lower extremities. Then, He was referred to our endocrinology department for clinical suspicion of CS. On physical examination, he had facial plethora, melanoderma, a muscle atrophy, purple abdominal striae and centripetal obesity (Figure 1).
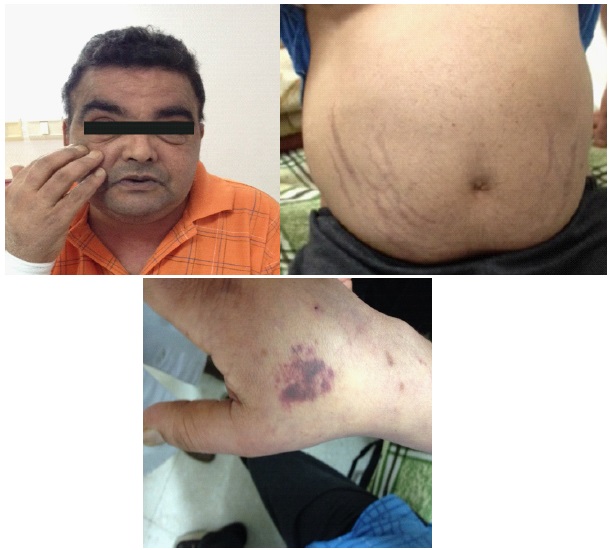
A laboratory examination revealed blood glucose level of 10 mmol/L, hypokalemia at 3.5 mmol/L and both high levels of cholesterol and low-density lipoprotein cholesterol at 5.88 and 3.7 mmol/l, respectively
His endocrinological workup revealed a plasma adrenocorticotropic hormone (ACTH) level of 219 ng/l, morning serum cortisol (MC) of 767 nmol/L, 24 h urine-free cortisol (UFC) of 887 ng, and unsuppressed cortisol after 1 mg overnight dexamethasone suppression test. Low-dose dexamethasone suppression testing (LDST) showed a plasma cortisol level of 586 nmol/L. High-dose dexamethasone suppression testing (HDST) was performed with an unsuppressed cortisol level of 434 nmol/L (< 50% suppression from baseline serum cortisol). The assessment of complications revealed osteopenia at the bone densitometry (T-score at−2.4) and gastritis due to helicobacter pylori infection at the fibroscopy with intestinal metaplasia treated medically.
The diagnosis of ectopic ACTH-dependent Cushing’s syndrome was established. Imaging tests were carried out to determine the source: Pituitary MRI showed no lesions. The abdominal and chest enhanced Computed Tomography (CT) showed a diffuse interstitial infiltrates syndrome with bilateral adrenal hyperplasia. No tumor was detected.
Meanwhile, the clinical situation did not improve. During hospitalization, his condition worsened, and he developed mental confusion with acute attack of paranoid delusion and hallucination improved by neuroleptic treatment, anxiolytic and anti-depressants. Osteopenia was complicated by rib fractures and vertebral marrow without impact to the spinal MRI. During his hospitalization, our patient presented a pulmonary embolism and recurrent fungal infections.
Based on the highly elevated cortisol production and inability to provide Mitotane, a treatment with a rapid acting somatostatin analogue (Octreotide) was started with a biological good response. There was a decrease in cortisol levels after the administration of octreotide from 855 nmol /l to 700 nmol/l in few days.
Subsequently, octreoscan was performed, showing a fixation in a suspect lung node in the middle lobe (Figure 2). A new reading of the CT scan report found the pulmonary nodule measuring 15 mm diameters in the right middle lobe, the same found in the Octreoscan.
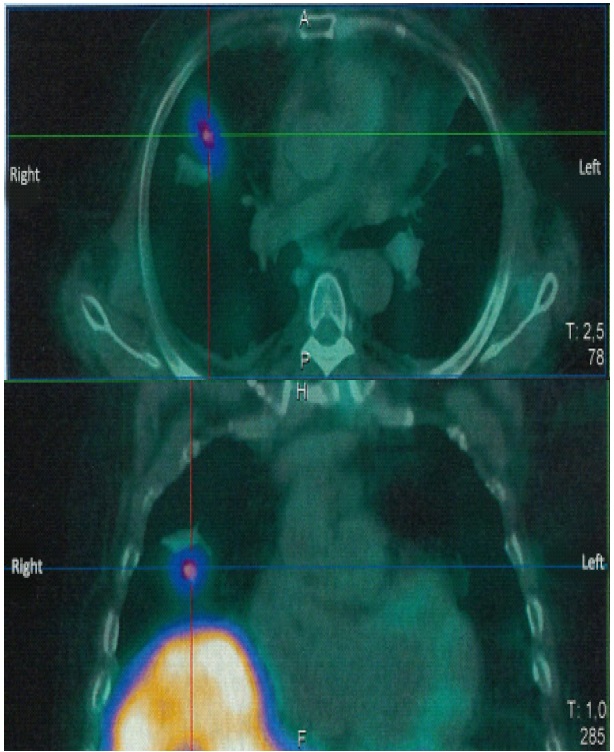
Our patient was operated on two weeks later with a right middle lobectomy, yet no nodule was identified on pathological examination. The patient developed a corticotropic deficiency two months later, and he was treated with hydrocortisone. The duration of follow-up was six years. No manifestations of CS were noted. Six years after his surgery, he still had no clinical features of hypercortisolemia (Figure 3). Biochemical workup was as follows: morning serum cortisol of 314 nmol/l, ACTH 31.6 ng/ml, and Low-Dose Dexamethasone Suppression Testing (LDST) showed a plasma cortisol level of 24 nmol/l.

In table 1, we summarized the evolution of cortisol and ACTH levels after our patient’s surgery. During treatment, his electrolyte imbalances were corrected. His diabetes and his hypertension were controlled better by a healthier lifestyle and only two drugs, respectively. His hemoglobin A1C and his fasting blood glucose were in normal range, [5.4-6%] and [4.4-5 mmol/l] respectively, without medical drugs. Currently, he is still being regularly monitoring for his retinopathy and nephropathy.
Table 1: Evolution of cortisol and ACTH levels after the surgery.
|
Before surgery |
1 day |
1 week |
2 weeks |
1 year |
2 years |
5 years |
6 years |
|
Morning serum Cortisol(nmol/l) |
700 |
324 |
298 |
80.7 |
84.75 |
192 |
284 |
314 |
|
ACTH (ng/l) |
Basic level |
219 |
- |
- |
- |
- |
- |
- |
31.6 |
|
DST |
63.3 |
- |
- |
- |
- |
- |
- |
7.25 |
ACTH: Adrenocorticotropic Hormone; DST: Dexamethasone Suppression Test; PO: Post-Operative.
Discussion
Ectopic ACTH syndrome accounts for 10–20% of ACTH-dependent Cushing cases [4]. This ectopic secretion is secondary to neuroendocrine tumors of varying size and localization. The following are tumor types generally associated with ectopic ACTH secretion: small cell lung carcinoma (SCLC), neuroendocrine tumor of the lung, small intestines, and carcinoid tumor of the pancreas. In roughly 10–20%, the primary tumor cannot be identified and the origin of the ectopic ACTH secretion remains unknown, generally described as occult tumors. The source should be established because the excision of the tumor can be curative [5,6]. In fact, small cell lung carcinoma and lung carcinoids cause half of cases [7] because it contains most of ACTH secreting spots.
In 12–19% of cases, the usual imaging fails to show any tumor as a cause of ectopic ACTH secretion, and tumors may appear later after several years of ECS diagnosis [8]. Therefore, specific imaging is often required, probably because of the small tumor size and the difficulty in tumor localization. CT and MRI failed to detect these tumors in 50% of cases, as was the case with our patient. Other tests such as high-resolution CT, scintigraphy, or PET scan can be utilized. Typical carcinoid tumors have high concentration of somatostatin receptors. Furthermore, octreotide tests can be useful [9]. Octreotide binds to SSTR-2 and SSTR-5 with high affinity. The expression of SSTR in bronchial carcinoid tumors has not been investigated [10]. This test has a sensitivity of 57% and positive predictive value of 79% and failed to identify tumors with negative somatostatin receptors [10]. FDG-PET scan identifies small metabolically active lesions with a sensitivity of 64% and positive predictive value of 53% for occult disease, as well as F-Fluorine-18-dixydroxyphenylalanine (F-DOPA) PET scan which is more appropriate for later stages in undifferentiated neuroendocrine tumors [9,11]. In our case, an ectopic ACTH syndrome was suspected, so we performed several imaging tests to find the tumor; but the abdominal and thoracic CT was all negative. However, Octreotide test showed a fixation in a suspect lung node in the middle lobe which was founded later after a new reading of the CT scan.
As for the treatment, the best way to treat a paraneoplastic Cushing’s syndrome is to go about the tumor itself. Surgery is curative in >80% of patients, when performed [12]. If radical resection is possible, this is the treatment of first choice. If radical surgery is not possible, systemic therapy, depending on the tumor type, can be administered [9,13].
In cases where localization of primary tumors cannot be achieved, therapy to decrease cortisolemia is essential with follow-up imaging to detect and resect tumors. For a SCLC, for example, chemotherapy can be used. There was no evidence of antiproliferative effect, but somatostatin analogs have been successfully used to treat paraneoplastic manifestations related to SCLC, as was the case with our patient. There are three different analogs: octreotide, lanreotide and pasireotide. Between octreotide and lanreotide, no significant difference in clinical activity has been found. If somatostatin analogs are given in the long-term, resistance can occur.
To decrease the high cortisol levels, inhibitors of glucocorticoid synthesis or action can be used. The available agents are: ketoconazole, metyrapone, etomidate, mitotane and mifepristone [14,15]. All these drugs reduce cortisol levels very rapidly. Bilateral adrenalectomy can be performed as a last resort if lifethreatening complications emerge.
Concerning the follow-up, there are two main factors that determine the prognosis in patients with an ACTH secreting tumor: cortisol level and tumor type. Patients with SLCL have the worst prognosis dying within 12 months of diagnosis, while patients with bronchial carcinoids have the best prognosis [8,16].
The second prognostic factor is the level of hypercortisolemia. The higher it is, the worse the prognosis. In patients with an occult tumor, the prognosis is generally better, provided that the hypercortisolemia is controlled [17-20].
Conclusion
This case illustrates the dilemma between the need for morphological diagnosis of the ectopic ACTH source and control of the life-threatening hypercortisolism. Indeed, ACTH secreting tumors can be very hard to detect, in such cases we should first aim to lower blood cortisol medically or through bilateral adrenalectomy to avoid Cushing’s complications. Such patients should then be followed up through imaging tests (CT, MRI, scintigraphy or PET) to detect the tumor and resect it, which constitutes the definitive treatment of these patients.
References
- Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, et al. The Diagnosis of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 1 mai 2008; 93: 1526‑40.
- Nieman LK. Cushing’s syndrome: update on signs, symptoms and biochemical screening. Eur J Endocrinol. 2015; 173: M33‑8.
- Valassi E, Franz H, Brue T, Feelders RA, Netea-Maier R, et al. Diagnostic tests for Cushing’s syndrome differ from published guidelines: data from ERCUSYN. Eur J Endocrinol. 2017; 176: 613-24.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, et al. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab. Août. 2005; 90: 4955‑62.
- Tani Y, Sugiyama T, Hirooka S, Izumiyama H, Hirata Y. Ectopic ACTH syndrome caused by bronchial carcinoid tumor indistinguishable from Cushing’s disease. Endocr J. 2010; 57: 679‑86.
- Zemskova MS, Gundabolu B, Sinaii N, Chen CC, Carrasquillo JA, et al. Utility of various functional and anatomic imaging modalities for detection of ectopic adrenocorticotropin-secreting tumors. J Clin Endocrinol Metab. mars 2010; 95: 1207‑19.
- Beuschlein F, Hammer GD. Ectopic pro-opiomelanocortin syndrome. Endocrinol Metab Clin North Am. Mars. 2002; 31: 191-234.
- Kenchaiah M, Hyer S. Cushing’s Syndrome due to Ectopic ACTH from Bronchial Carcinoid: A Case Report and Review. Case Rep Endocrinol. 2012; 2012: 215038.
- Meftah A, Moumen A, Massine El Hammoumi M, Hajhouji S, et al. [Paraneoplastic Cushing’s syndrome, a real diagnostic and therapeutic challenge: A case report and literature review]. Rev Med Interne. 2015; 36: 843‑7.
- Menezes Nunes J, Pinho E, Camões I, Maciel J, Cabral Bastos P, et al. A challenging case of an ectopic cushing syndrome. Case Rep Med. 2014; 2014: 413136.
- Li W-Y, Liu X-D, Li W-N, Dong S-Y, Qu X-H, et al. Paraneoplastic Cushing’s syndrome associated with bronchopulmonary carcinoid tumor in youth: A case report and review of the literature. Oncol Lett. Juill. 2016; 12: 69‑72.
- Han SY, Kim BH, Jang HR, Kim WJ, Jeon YK, et al. Ectopic ACTH syndrome caused by pulmonary carcinoid tumor mimicking long-standing sclerosing hemangioma. Korean J Intern Med. Juill. 2016; 31: 794‑7.
- Molina Garrido MJ, Guillén Ponce C, Maciá Escalante S, Pons Sanz V, Carrato Mena A. Cushing’s paraneoplastic syndrome as first manifestation of an adenocarcinoma of unknown origin. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mex. Août. 2006; 8: 621‑3.
- Aljassem G, Aljasem H. Case report: Ectopic Cushing’s syndrome in a young male with hidden lung carcinoid tumor. Int J Surg Case Rep. 2018; 42: 13‑6.
- Zhang H-Y, Zhao J. Ectopic Cushing syndrome in small cell lung cancer: A case report and literature review. Thorac Cancer. 2017; 8: 114‑7.
- Deldycke A, Haenebalcke C, Taes Y. Paraneoplastic Cushing syndrome, case-series and review of the literature. Acta Clin Belg. Août. 2018; 73: 298‑304.
- Witek P, Witek J, Zieliński G, Podgajny Z, Kamiński G. Ectopic Cushing’s syndrome in light of modern diagnostic techniques and treatment options. Neuro Endocrinol Lett. 2015; 36: 201‑8.
- Kanaji N, Watanabe N, Kita N, Bandoh S, Tadokoro A, et al. Paraneoplastic syndromes associated with lung cancer. World J Clin Oncol. 2014; 5: 197‑223.
- Nagy-Mignotte H, Shestaeva O, Vignoud L, Guillem P, Ruckly S, et al. Prognostic impact of paraneoplastic cushing’s syndrome in small-cell lung cancer. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. Avr. 2014; 9: 497‑505.
- Davi’ MV, Cosaro E, Piacentini S, Reimondo G, Albiger N, et al. Prognostic factors in ectopic Cushing’s syndrome due to neuroendocrine tumors: A multicenter study. Eur J Endocrinol. avr 2017; 176: 451‑9.