
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Re-emergence of a rare syndrome: Mauriac syndrome
Sattik Siddhanta1*; Soumik Goswami2; Nilanjan Sengupta3; Pranab Kumar Sahana4
1 Assistant Professor, Department of Medicine, IPGMER and SSKM Hospital, Kolkata, West Bengal, India.
2 Tutor, Department of Endocrinology, NRS Medical College, Kolkata, West Bengal, India.
3 Professor & Head, Department of Endocrinology, NRS Medical College, Kolkata, West Bengal, India.
4 Associate Professor, Department of Endocrinology, NRS Medical College, Kolkata, West Bengal, India.
*Corresponding Author: Sattik Siddhanta
Assistant Professor, Department of Medicine,
IPGMER and SSKM Hospital, Kolkata, West Bengal,
India.
Email: drcalmed@gmail.com
Received : Sep 16, 2021
Accepted : Oct 21, 2021
Published : Oct 28, 2021
Archived : www.jcimcr.org
Copyright : © Siddhanta S (2021).
Citation: Siddhanta S, Goswami S, Sengupta N, Sahana PK. Re-emergence of a rare syndrome: Mauriac syndrome. J Clin Images Med Case Rep. 2021; 2(5): 1387.
Introduction
Mauriac syndrome (MS) is one of the rare complications of poorly controlled Type 1 Diabetes Mellitus (T1DM). It is characterized by poor glycemic control, short stature hepatomegaly, Cushingoid features and delayed puberty [1]. Since the introduction and easy availability of longer acting insulin and insulin analogues, cases of Mauriac syndrome are hardly reported now [2]. This syndrome is more frequently encountered in children and adolescents with poor glycemic control and increases susceptibility of micro vascular complications of Diabetes like diabetic retinopathy and diabetic nephropathy. It is the most common cause of hepatic dysfunction in children and adolescents with Type 1 diabetes mellitus [3]. A classical case of Mauriac syndrome (MS) in an adolescent girl of uncontrolled Type 1 Diabetes Mellitus is being presented.
Case presentation
A 15 year old girl, born out of non consanguineous marriage and belonging to low socio economic strata was referred for poor glycemic control. She was diagnosed as Type 1 Diabetes Mellitus two years ago following an episode of diabetic ketoacidosis. Since then, she was on premixed insulin (30% short acting and 70% NPH insulin) in twice daily doses (42 units/day at present). However, due to poor compliance to therapy and ignorance about frequent glucose monitoring, she had undergone multiple episodes of hospitalizations for Diabetic Ketoacidosis and severe hypoglycemic convulsions despite repeated education regarding titration of insulin dosage and schedule. Her parents were extremely anxious and apprehensive due to her poor glycemic control. Recently, for the last six months she also complained of abdominal heaviness and a dull aching abdominal pain which is hampering her activity of daily living and compromising her quality of life significantly. Other history was noncontributory except that she did not attained menarche yet. She was immunized in accordance with her age. Family history was non contributory.
On examination, anthropometry revealed she was found to be significantly short for her age (133 cms, <3rd percentile), Height SDS:- 3.5, Height age- 10.5 years. Her weight was 33 kgs (<3rd percentile), Weight SDS:- 1.81, Weight age was 10 years. She had a Body Mass Index (BMI) of 19.88 kg/m2 . She had moon facies (Figure 1) but no icterus, edema or goiter. Her vital signs were normal and her Sexual Maturity reading (SMR) was B3P1. Abdominal examination revealed massive firm nontender hepatomegaly with a liver span of 18 cms. However, there was neither any free fluid in the abdomen nor any evidence of hepatocellular failure. She had lipohypertrophy at the injection sites suggestive of poor injection technique. Other systemic examination including ophthalmoscopy was normal.

Following admission, her random blood glucose levels was 406 mg/dl but urinary ketone bodies were negative. She was started on multiple subcutaneous insulin injections – basal bolus insulin regimen with frequent monitoring of blood glucose with a glucometer at least four times a day and titration of insulin doses as per the capillary blood glucose readings. Routine hematological and biochemical investigations revealed a normal complete hemogram. (Hb-12 gm%, Total Leucocytes Count (TLC)-6300/mm3 , N66L29E2M2B0, Pltcount - 3.7 lacs/ mm3 ). Her glycated hemoglobin was 13.2% (normal: 4.2-6.4%) on presentation suggestive of poor glycemic control. Her renal function tests and liver function tests were within normal limits. Urinalysis showed evidence of glycosuria and mild proteinuria but no ketonuria. (Urine RE/ME-glucose+++, protein+, ketonesabsent). Liver Function Tests (LFT) showed evidence of transaminitis with preserved synthetic function. [Bilirubin (Total) 1.1, Bilirubin (Conjugated)-0.4, Total Protein - 7.2, Albumin - 4.1, SGPT - 466U/L(Normal: 5-45 U/L), SGOT-502 U/L (Normal: 5-45 U/L), ALP 116]. Prothrombin time/INR was within normal limits. Her Lipid Profile revealed evidence of atherogenic dyslipidemia [Total Cholesterol‑394 mg/dl (125-189), Triglycerides ‑ 194 mg/ dl(28-85), Low Density Lipoprotein Cholesterol(LDL-C) ‑131 mg/ dl(63-129), High Density Lipoprotein (HDL-C)‑ 23 mg/dl (38-74)].
Her Fasting Plasma Glucose (FPG) was 353 mg/dl, and postprandial plasma glucose (PPBG) 480 mg/dl; serum C-peptide (fasting) was <0.1 ng/ml and serum C-peptide (post mixed meal) was <1.19 ng/ml; she had very low (44.4 ng/ml) IGF-1 (somatomedin C) but serum cortisol was 7.5 μg/dl (normal); however glutamic acid decarboxylase (GAD-65) antibody was 184 IU/ml(positive) and HbA1c was 13.2%. Islet-cell antibody and anti-insulin antibody results were not significant. Follicle stimulating hormone level was 3.2 mIU/ml (normal 1.27– 11.95); luteinizing hormone was 0.46 mIU/ml (normal 1.14–8.75); serum Estradiol (E2) of 35.44 pg/ml was low and prolactin was 15.58 ng/ml (normal 3.46–19.40). She had negative report of serum antinuclear antibody, anti-double stranded deoxyribonucleic acid and tissue transglutaminase IgA. Serum vitamin D was 14.8 ng/ml (normal 30–47), and serum vitamin B12 was 488 pg/ml (normal 191–663). Her bone age was delayed (11 years). Imaging in the form of Ultrasound of abdomen confirmed massive hepatomegaly with increased hepatic parenchymal echogenicity. There was no other organomegaly or presence of free fluid in abdomen.
Differential diagnosis
At this juncture, we had the following differential diagnosis
in mind:
a. Non Alcoholoic Fatty Liver Disease (NAFLD)
b. Glycogen Hepatopathy
c. Autoimmune Hepatitis
d. Viral Hepatitis
e. Metabolic (Alpha 1 antitrypsin deficiency, Wilsons disease).
To streamline the aetiology, viral markers (Hbs Ag & Anti HCV) sent were negative. Anti nuclear antibody (ANA), Anti LKM antibody and Anti Sm antibody all were found to be negative too. alpha-1-antitrypsin and serum ceruloplasmin levels were within normal limits. Hence to differentiate between non alcoholic fatty liver disease and glycogen hepatopathy, a transcutaneous ultrasound guided liver biopsy was performed. Liver biopsy revealed findings consistent with diffuse glycogen-rich cytoplasm (Figure 2) and glycogenated nuclei of hepatocytes without any steatosis, fibrosis or inflammation. PAS staining was positive for glycogen accumulation which abolished with Diastase pretreatment thereby conforming a pathological diagnosis of Glycogen Hepatopathy.
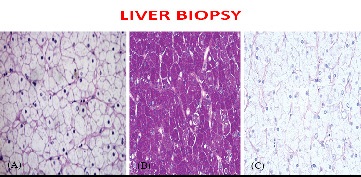
Based on the clinical features of short stature, cushingoid habitus, massive hepatomegaly, delayed puberty with biochemical evidence of raised liver ezymes and dyslipidemia and glycogen laden hepatocytes, A provisional diagnosis of Mauriac Syndrome in a poorly controlled Type 1 Diabetes mellitus adolescent female was made.
Following discharge, she was maintained on s/c basal bolus insulin regimen, motivated for frequent self monitoring of blood glucose levels at periodic intervals, trained about proper insulin injection adminstration technique, sick day rules and frequent rotation of injection sites. Cholecalciferol granules (60000 IU) was given once weekly for eight weeks. At 6 months of follow up she had achieved optimization of glycemic control without any episode of hypoglycemia or Diabetic Ketoacidosis. There has been a significant reduction of her hepatomegaly with substantial improvement of abdominal pain. She gained a height of 5 cms and weight of 3 kgs and experienced menarche. Investigations revealed a significant reduction of glycated hemoglobin and marked improvement of her lipid profile with normalisation of transaminase levels.
Discussion
In 1930, Mauriac first described a syndrome in patients with uncontrolled Type 1 Diabetes Mellitus (T1DM) presenting with clinical features of growth failure, pubertal delay, hepatomegaly and protruded abdomen [4]. Since the advent of the long-and intermediate-acting insulins and insulin analogues, the various clinical manifestations of Mauriac syndrome are rarely encountered. Most of the cases usually occur in adolescent age group with an equal gender ratio.
Mauriac Syndrome (MS) is further classified into two subgroups depending upon the presence or absence of obesity. In the obese variety of the syndrome, poor glycemic control and glycemic variability results in severe and wide fluctuations between hyperglycemia and hypoglycaemia, suggestive of a pattern of over- and under insulinisation of glucose, respectively. In the non-obese variety of the syndrome, patients were inadequately insulinised without any history of alternating hypoglycaemia and ketoacidosis [5]. The pathogenesis of this syndrome is unknown but is thought to be multifactorial. Inadequate glucose to the tissues, decreased IGF‑1 and growth hormone levels, hypercortisolism, and resistant/defective hormone receptor action may lead to stunted growth and delayed puberty [6]. MacDonald et al. [7] Discovered a mutation in the catalytic subunit of liver glycogen phosphorylase kinase in a patient with Mauriac syndrome who developed growth failure and massive hepatomegaly. In their patient, PHKG2 G→A mutation in exon 9 was identified for excess glycogen accumulation in liver cells. Glycogen phosphorylase kinase activates glycogen phosphorylase, the enzyme that catalyses the first step in glycogen breakdown. This case proves that the effect of a mutant enzyme of glycogen metabolism can combine with hyperglycaemia to directly hyper-inhibit glycogen phosphorylase, in turn blocking glycogenolysis causing the massive liver in Mauriac disease. Thus, neither hyperglycaemia alone nor a mutated enzyme alone are sufficient to cause this syndrome: a mutant enzyme of glycogen metabolism combined with chronic hyperglycaemia can cause the syndrome [7]. Hepatomegaly developed because of glycogen deposited in the liver due to periods of supraphysiological levels of insulin. Blood glucose passively enters the hepatocytes in which glycogen synthesis is promoted by high cytoplasmic glucose concentration reliant on the presence of insulin. Then glycogen is deposited within the hepatocytes as a result of a vicious cycle of hyperglycaemia and insulin treatment [8]. Poor glycaemic control due to hypoinsulinaemia leads to lipolysis and ketones production. Ketosis activates cortisol synthesis promoting the release of fatty acids and hyperglycaemia [9].A transcutaneous ultrasound guided Liver biopsy is the gold standard for confirmation of clinical diagnosis of Mauriac Syndrome. Histolopathologic features are characterized by large, swollen, glycogen-laden hepatocytes and glycogenated nuclei without significant fatty change, inflammation, lobular spotty necrosis or fibrosis [8].A high index of suspicion of Mauriac Syndrome should be kept in mind when any patient with poorly controlled diabetes mellitus presents with hepatomegaly, elevated transaminases, dyslipidemia, growth and pubertal delay with Cushingoid features. An early and durable glycemic control not leads to improvement in liver function tests and lipid profile but also results in improvement of hepatomegaly, increase in anthropometric parameters and onset of puberty.
In this case, poor compliance to therapy because of illiteracy and ignorance was the primary cause of poorly-controlled diabetes. In addition, prior to her admission under our care poor socio economic background compelled her to be put on premix insulin regimen. After achieving the better glycaemic control with multiple subcutaneous insulin injection therapy, the patient achieved excellent glycemic control and recovered from the complications with remarkable improvement in growth and development and resolution of hepatomegaly was observed too.
Conclusion
To conclude, Mauriac syndrome though a very rarely encountered complication of poorly-controlled T1DM in modern days, yet it is quite frequent in rural setting in India where premix insulin are still rampantly used over modern long acting insulin analogues due to socio economic reasons. The clinician must have a high index of suspicion so that timely intervention can yield proper growth and pubertal maturation. This can be accomplished by ensuring compliance with insulin therapy (either insulin pumps or multiple subcutaneous insulin injections), preferably long acting insulins and rapid acting insulin analogues in proper dosage, and providing supportive treatment, nutritional support and good follow-up care.
Take home messages
• Mauriac syndrome though a rare complication of poorly controlled type1 diabetes in adolescence and young adults with the advent of modern insulin analogues and intensive insulin regimens in modern days, yet it is quite frequently encountered in rural population.
• This syndrome is manifested clinically as short stature, hepatomegaly, pubertal delay, Cushingoid facies and biochemically as elevated transaminases and dyslipidemia in the background of poorly controlled Type 1 Diabetes Mellitus.
• A thorough history taking, meticulous systemic examination supplemented by necessary biochemical and radiological investigations remain of paramount importance for diagnosis.
• A transcutaneous ultrasound guided liver biopsy remain the gold standard for diagnosis of Mauriac Syndrome.
• Timely intervention ensuring compliance with modern insulin therapy and intensive insulin regimens, supportive treatment including nutritional and psychological support, regular physical exercise and good follow-up care can help in recovery from Mauriac syndrome.
References
- Maia FF, Araújo LR. Pancreas transplantation in Mauriac Syndrome:Clinical and biochemical parameters after one year follow up. ArqBras Endocrinol Metabol. 2005; 49: 455‑9.
- Alemzadeh R, Ali O. Diabetes mellitus. In: Kliegman RM Stanton BF, Schor NF, Geme JW, Behrman RE, editors. Nelson Textbook of Pediatrics. 19th ed. Philadelphia: Elsevier. 2012; 1968-97.
- Flotats Bastardas M, Miserachs Barba M, Ricart Cumeras A, et al. Hepatomegaly due to glycogen storage disease and type 1 diabetesmellitus. An Pediatr (Barc) 2007; 67: 157-60.
- Mauriac P. Gros ventre, hepatomegalie, troubles de croissance chez les enfants diabetiques traits depuis plusiers annee par l’insuline. Gaz Hebd Med Bordeaux. 1930; 26: 402–10.
- Rosenbloom AL, Silverstein JH. Diabetes in the child and adolescent. In Pediatric endocrinology. Lifshitz F (ed). New York, NY, USA: Marcel Dekker. 2003; 621–2.
- Lee RG, Bode HH. Stunted growth and hepatomegaly in diabetes mellitus. J Pediatr. 1977; 91: 82‑4.
- MacDonald MJ, et al. Discovery of a genetic meta¬bolic cause for Mauriac syndrome in type 1 diabetes. Diabetes 2016; 65: 2051–9.
- Torbenson M, Chen YY, Brunt E, Cummings OW, Gottfried M, et al. Glycogenic hepatopathy: An underrecognized hepatic complication of diabetes mellitus. Am J Surg Pathol. 2006; 30: 508-13.
- Pigui A, Montembault S, Bonte E, Hardin JM, Ink O. Voluminous hepatomegaly in a young diabetic patient. Gastroenterol Clin Biol. 2003; 27: 1038‑40.