
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
A rare entity of malignant insulinoma diagnosed after 30 years of clinical manifestation of hypoglycemia: A case report and literature review
Saleem Abdel Backi1 ; Toufic Saber1 ; Ziad el Rassi2 *
1 Department of General Surgery, Saint Georges Hospital University Medical Center, Beirut, University of Balamand, Lebanon. 2 Head of General Surgery Department, Saint Georges Hospital University Medical Center, Beirut, University of Balamand, Lebanon
*Corresponding Author: Ziad el Rassi
Professor of General Surgery, Head of General surgery
Department, Saint Georges Hospital University Medical
Center, Beirut, University of Balamand, Lebanon.
Email: ziadelrassi@gmail.com
Received : Sep 27, 2021
Accepted : Nov 17, 2021
Published : Nov 24, 2021
Archived : www.jcimcr.org
Copyright : © Rassi Z (2021).
Abstract
Malignant insulinomas, a rare life threatening pathology, exists in literature as an entity that constitutes 10% of all insulinomas and often present as multi-centric macro nodules with multiple lymph nodes or liver metastases before diagnosis. We report a rather rare case of a 68 year old male with a 30 years history of uninvestigated severe hypoglycemic attacks that improved on glucose intake. Blood tests showed a decreased value of glycemia (45 mg/dL) associated with increased insulin level (54 μU/ml) and an increased glycemia/ insulinemia ratio of 0.83 supporting the diagnosis of insulinoma. Abdominal CT showed a 4 cm mass localized in the head of the pancreas with atrophic body and tail, no signs of distant metastatic disease. A concomitant diagnosis of primary hyperparathyroidism raised, based on elevation of calcium associated and high level of PTH. The coexistence of the two endocrinopathies suggested the presence of type 1 multiple endocrine neoplasia (MEN I). Based on the workup suggesting a benign insulinoma with no signs of metastatic disease, co-existing with debilitating symptoms of hypoglycemia, pancreatectomy with lymph node dissection was performed. Histo-pathological examination returned surprisingly positive for malignant neuro-endocrine tumor with positive lymph nodes. In that domain, we summarized the literature discussion of neuroendocrine tumors, elaborating on malignant insulinoma diagnosis and management. Furthermore, what our article is trying to lay upon existing literature is a case of a long standing existent MEN 1 malignant insulinoma manifesting as a remarkably slow progressive disease of 30 years’ timeline versus a less likely chance of a transformation from benign insulinoma to malignant
Keywords: neuroendocrine neoplasm; pancreatectomy; MENI; malignant insulinoma; chronic hypoglycemia.
Citation: Backi SA, Saber T, Rassi Z. A rare entity of malignant insulinoma diagnosed after 30 years of clinical manifestation of hypoglycemia: A case report and literature review. J Clin Images Med Case Rep. 2021; 2(6): 1423.
Introduction
Insulinoma, as first described in 1927, is a functioning neuroendocrine neoplasm that produces excessive amounts of insulin causing symptomatic hypoglycemia prompting patients to seek medical attention early during the course of their disease. This novel entity surfaced after a patient with symptoms of hypoglycemia was found to have metastatic primary pancreatic tumor, the extracts of which induced hypoglycemia in in-vivo animal trials [1]. Although insulinomas being the most common islet cell tumors that localized almost exclusively within the pancreas (over 99% of them and less than 1% in ectopic pancreas tissue), it is a rare life threatening pathology with an annual incidence of 1-3 per 1 million persons per year. There is a slight female predominance, with 60% of new cases diagnosed in women and median age at presentation of 47 years [2]. Most insulinomas are sporadic (90%), solitary (83-92%) and benign (90%), typically hypervascular tumors, measuring in 90% of cases less than 2 cm. While benign insulinomas were well elaborated upon in numerous studies that have assessed its epidemiology, treatment and prognosis, malignant insulinoma still emerge as a poorly understood entity. In that direction, the few case series assessing malignant insulinoma, have looked only at distant metastatic disease, and hence data about the frequency and prognosis of other stages are still underreported [3,4]. Also very few studies have provided meaningful evidence regarding malignant insulinoma characteristics, trends and outcomes, but it is accepted that overall survival for malignant insulinoma is significantly worse than for benign disease [3].
Only 5.8-15% of insulinomas are multicentric macro adenomas (measuring over 2.5 cm), at a greater risk of harboring malignant lesions, with higher recurrence rates and unlike curative resection [3,5,6]. Until now, the World Health Organization classifies an insulinoma as malignant solely based on the presence of metastases, most commonly to lymph nodes followed by liver [7]. Tumor size ≥2 cm, CK (cytokeratin) 19 status and tumor staging and grading (Ki 67 labeling > 2%) are reported as predictors of malignant metastatic disease [8-10]. Patients diagnosed with insulinoma and distant metastases to liver, bone and lymph node have been found to have a median survival of < 2 years [11]. Approximately 50% of malignant insulinomas (half of them with disseminated hepatic metastases) and 7.6-16% of all insulinomas are associated with type 1 multiple endocrine neoplasia (MEN1). Insulinomas represent the second most common type presentation (20-45%) of MEN 1 after primary hyperparathyroidism (almost always present-95%), having an unfavorable outcome and higher recurrence rates [12].
Worth mentioning and high on the list of acquainting with the disease is the thoroughly described struggle regarding its different aspects from the unspecific clinical presentation, to diagnostic workup, choice of surgical intervention, and pathological classification [13].
As to the management of both, complete surgical resection of an insulinoma, once localized, is the treatment of choice to cure the debilitating hypoglycemic symptoms associated with insulinoma [14,15]. Worth of mention is that surgery can be challenging in the presence of malignant disease as these patients can present with unresectable lesions.
Under malignant insulinoma and due to the rarity of the condition, we present hereby a case of a 68-year-old man with long standing history (>30 years) of uninvestigated hypoglycemia who presented to our institution with severe hypoglycemia and was investigated to a pancreatic head tumor and malignant histo-pathological examination; An unprecedented case of a giant (by size), long standing insulinoma, with the full blown symptomatology, associated with MEN I which turned out to be malignant upon resection.
Case report
A 68 years old man presented to our Emergency Department (ED) for few hours history of dysarthria and generalized fatigue. He only reported a 30 years standing history of hypoglycemia, uninvestigated, and managed conservatively, compatible with whipple’s triad of diagnosing insulinoma. The patient has no other significant past medical history or surgical history, did not take any medications at home, with no known allergy to a drug or food and negative smoking history. He had a negative family history for pancreatic or endocrine tumors.
In ED, he was hypertensive (SBP 18/DBP 10), otherwise stable vital signs; however he had a significantly low non-fasting blood glucose level of 45 mg/dL. A brain MRI was done through ED to rule out an overlying cerebrovascular accident returned negative. Patient was admitted for regulation of hypoglycemia with continuous intravenous (IV) dextrose and further diagnostic investigations.
Further blood workup was significant for WBC count of 17,000 but negative neutrophilic shift, low HbA1c of 3.5 and high serum insulin levels of 54 μU/ml. Surprisingly patient had elevated Calcium levels of 11.3 mg/dl with Parathyroid hormone of 160 pg/ml, and low vitamin D of 11.3.
A CT-scan of abdomen and pelvis with IV contrast done for investigating any underlying etiology revealed irregularly enlarged pancreatic head and uncinate process (4.3 X 3.6 cm) with severe atrophy of body and tail, dilated main pancreatic duct of 9 mm and few adjacent enlarged lymph nodes (up to 11 mm); findings compatible with a pancreatic mass however no contact of the mass with IVC, SMV, SMA, celiac trunk. No common bile duct dilation. No signs of distant metastasis. Also a large Staghorn calculus found occupying the right renal pelvis. Abdomen MRI/MRCP confirmed the CT-scan findings (Refer to Figures 1-3).
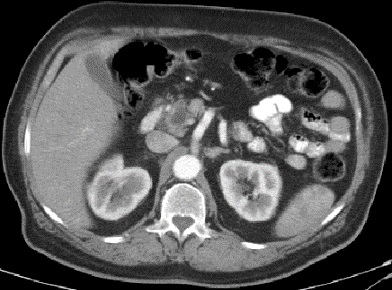
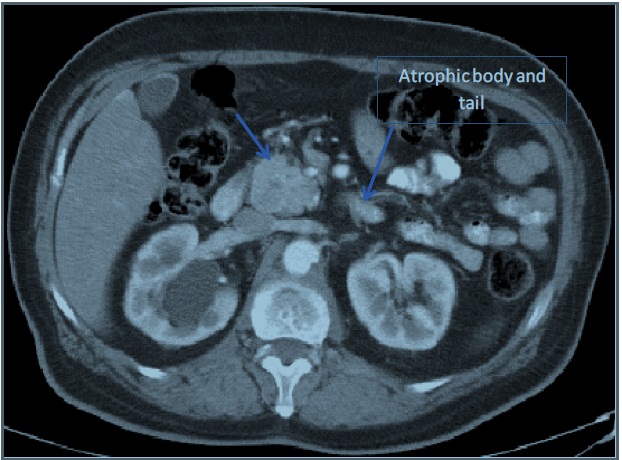
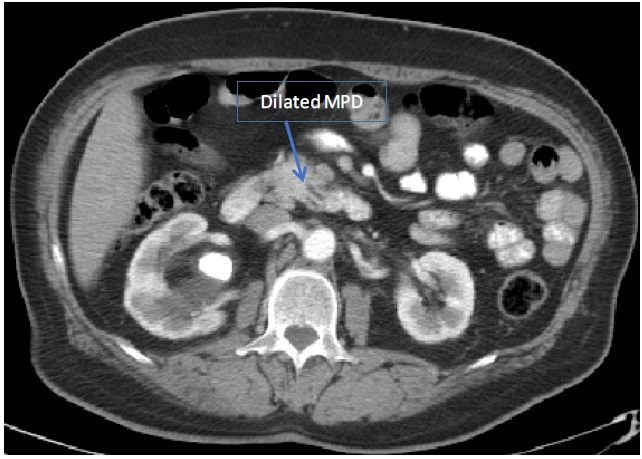
Having primary hyperparathyroidism and a pancreatic mass clinically correlated to being an insulinoma, patient was diagnosed with MEN I. However no genetic studies for diagnosis of MENI were done due to insufficient funds. Of importance to mention is negative workup for any co-existing pituitary gland pathology on MRI, and an adrenocorticotropic hormone profile within normal ranges. The need for surgical intervention and resection of the pancreatic mass was discussed with patient and operation scheduled.
In operation, no signs of metastatic disease noted, a total pancreatectomy was done with duodenectomy (classical, nonpylorus preserving), cholecystectomy, extensive lymph node dissection and re-establishment of the alimentary tract with roux en Y gastro-jejunostomy and hepatico-jejunostomy.
Post operatively, patient was clinically and hemo-dynamically stable, uncomplicated stay in hospital with a remarkable appropriate control of blood glucose levels off inuslin. A control CT- scan done Day 7 post op showed intact anastomosis with no signs of leak or any intra-abdominal collection or rather active processes. Tri-phasic diabetic high caloric diet initiated and progressed accordingly with good patient tolerance. Patient was discharged on day 10 post-op with the plan of further investigating as outpatient the suspicion of parathyroid pathology due to the revealed primary hyperparathyroidism.
Histo-pathological examination report A well differentiated pancreatic neuro-endocrine tumor of the pancreatic head Grade G2/G3 (Ki-67 labeling index: 8%) with strong diffuse expression of Synaptophysin, CD 56 and Chromogranin A, metastatic to lymph nodes (16 out of 24 peripancreatic lymph nodes were positive). No evidence of lymphovascular or perineural invasion. All surgical margins were clear of neoplasm.
Pathology stage: pT2N1 (III) by AJCC.
Discussion
In this article, we reported a case of uninvestigated 30 years history of symptomatic hypoglycemia. MENI insulinoma was evident on biochemical workup that showed primary hyperparathyroidism with severely low blood glucose levels along with a low HbA1c and elevated serum insulin levels. In this direction, diagnostic imaging done revealed a pancreatic head tumor, clinically correlated to an insulinoma and presenting itself as chronically existing due to atrophy of pancreatic body and tail, as a part of pancreatic tissue accommodation to a long standing, pancreatic head positioned, hyper-functioning tumor.
Insulinoma is a beta-cell-derived NET of pancreas responsible for unregulated secretion of insulin. Compared to other entero-pancreatic tumors such as gastrinomas or glucagonomas, which are frequently malignant (46-60%), most of insulinomas are benign. Malignant insulinoma is a rare pancreatic neuroendocrine tumor that represents 5.8 to 15 % of all insulinomas [3,5,6]. While this neoplasm is slow growing, it represents a challenge for the managing physician for diagnosis and therapeutic approach and hence studying trends and factors impacting survival is important to provide more insight into this disease [3,16]. Diagnosis must be established through identifying presence of local invasion or metastases (most frequently in the peri-pancreatic lymph nodes and liver). Then moving on to unregulated excessive secretion of insulin and pro-insulin-related products in which the tumor presents itself as severe hypoglycemia and even hypoglycemic coma, which is a life-threatening condition. The average period of hypoglycemic episodes before diagnosis of insulinoma is established is generally 12-18 months [4]. In our case, the diagnosis of insulinoma came after 30 years history of symptomatic hypoglycemia that increased in severity and frequency few days in advance. Biochemical diagnosis of insulinoma was established, confirmed by imaging CT-scan and MRI showing a resectable pancreatic head tumor with atrophic body and tail.
Despite being slow-growing, some neuroendocrine tumors of may show an aggressive behavior and therefore identification of factors able to predict their natural history is important in order to appropriately approach an insulinoma. Recently, WHO adopted a new classification of endocrine tumors of the pancreas based on clinical and pathological criteria such as: tumor size, local invasion, necrosis, structural atypia with prevalence of broad solid areas, cellular atypia with high nuclear cytoplasmic ratio, irregular distribution of chromatin, number of mitoses/10 HPF, Ki 67 positive tumor cells, peri-neural and angio-invasion, tumor cells with immunoreactivity to neuroendocrine markers (synaptophysin, chromogranin A, insulin, proinsulin detected by immunohistochemistry), nuclear p53 protein accumulation [8- 10]. According to this classification, neuroendocrine pancreatic tumors differs between well-differentiated endocrine tumors (benign or low-grade malignant), well-differentiated endocrine carcinoma and poorly-differentiated endocrine carcinoma. Ki 67 is an index of proliferative status, suggesting a more aggressive behavior when present in more than 2% of tumoral cells [8-10], as was reported in the case of our patient. Still, the diagnosis of neuroendocrine carcinoma may be established only in the presence of metastases and/or invasiveness [4], with no conclusive and well-structured histological criteria or histochemical markers that reliably predict biological behavior.
Patients with insulinoma usually suffer from neuro-glycopenic symptoms (e.g. confusion, behavioral changes, loss of orientation) resulting from chronic inadequate supply of glucose brain cells leading to disrupted neuronal function. In this direction, pancreatic insulinoma remains a diagnostic dilemma as it is typically diagnosed after less than 1.5 years following the onset of symptoms and in many cases might be misdiagnosed as neurological or psychiatric disorders, leading to unnecessary investigations or unrelated psychiatric treatment. The classic hallmark on insulinoma is the whipple triad that consists of symptoms hypoglycemia (especially when fasting or exercising), documented low blood sugar (<50 mg/dl) at the symptoms are present and reversal of symptoms by glucose administration [3]. This has been evident in our case where all the components of this triad were acknowledged along with hypoglycemic symptomatology upon patient presentation. Another definitive clinically diagnostic modality of insulinoma is a 72-hour supervised fasting test where despite hypoglycemia and absence of serum sulfonylurea, there still exists an inappropriate rise in the levels of insulin (≥6 μU/mL) and C-peptide (≥0.2 nmol/L) [17].
Furthermore, in approaching neuroendocrine tumors, the possibility of MEN should always be taken into consideration and investigated as part of the comprehensive workup. MEN is a group of heritable syndromes characterized by the presence of benign or malignant tumors in endocrine tissues, and co-existence of two MEN-related endocrinopathies suggests the diagnosis of MEN. However final confirmation could be only achieved by genetic analysis. In our case, malignant insulinoma and primary hyperparathyroidism were two co-existing pathologies suggesting MEN 1. MEN 1 by definition, is an autosomal dominant inherited syndrome, and the gene responsible for developing disease is a tumor suppression gene called MENIN. Clinical entities described to be associated as MEN 1 are: Hyperparathyroidism (95%), enteropancreatic tumors (30-80%) that are mostly benign, pituitary adenomas (most frequent prolactinoma, followed by growth hormone secreting tumors; 20-25%), carcinoid tumors (20%), adrenal adenomas (40%) and subcutaneous lipomas (30%) [18].
Keeping in mind the hypercalcemia found in our patient that was biochemically attached to primary hyperparathyroidism, a great controversy exists in attaining surgical resection if a parathyroid pathology is to be identified. Although surgery is approved of to be the treatment of choice of hyperparathyroidism in MEN 1, it is still debatable in the presence of associated malignant enteropancreatic tumors. In such cases it may only be considered in non-progressive enteropancreatic tumors [19].
After establishing a diagnosis of insulinoma, the need to localize the tumor, as the corner stone of achieving surgical resection, urges the use of different imaging modalities based on availability of the technique and radiological skills. Non-invasive imaging modalities include US, CT scan, magnetic resonance imaging, Ga 68 DOTA-Pet, and positron emission tomography [20]. For how the above-mentioned localization modalities should be sequenced, no predefined algorithm is established and choice is solely based on regular clinical practice. In our case, a CT scan was done initially that localized the tumor to pancreatic head, along with staging. A further abdomen MRI/MRCP attained that confirmed the finding. Other invasive techniques can be introduced if the conventional non-invasive modalities fails, including Endoscopic Ultrasound (EUS), Selective Arterial Calcium Stimulation Test (SACST) and percutaneous transhepatic portal pancreatic venous sampling (PTVS to sample the blood for detecting insulin levels), all of which are highly sensitive however exist with high risk of complications up to 10%, excluding from this EUS which, depending on the operator, can exhibits a sensitivity up to 93% for detection of the tumor [21]. Laparoscopic exploration with intraoperative ultrasound (IOUS) is also being considered nowadays as Grover et al. called IOUS to be equivalent to SACST and PTVS for the localization of insulinoma [22].
After localization of the tumor, complete surgical resection also referred to as enucleation remains the gold standard for treating insulinoma, owing to the fact that the vast majority of cases exist as solitary and benign tumors. For larger or more extensive tumors, resection of a part of the pancreas through spleen preserving distal pancreatectomy or Whipple procedure is to be achieved. For those unfit for surgery or when tumor resection is unsuccessful, implementation of both somatostatin analogs (to which only 50% of patients respond) and α-interferon as diazoxide, can be used to inhibit the release of insulin [23].
For malignant insulinomas, although no general consensus delineating the optimal management approach, owing to the rarity of the disease, the decision should be taken by a multidisciplinary team, and despite surgical excision being the only curative option, it is rarely possible [23]. However, surgery is always to be considered the treatment of choice in a surgically fit patient, even in presence of hepatic metastases as long as least 90% of tumoral tissue can be removed [24,25]; these special cases with resectable liver metastasis has showed 50% remission rate at three years a significantly higher five year survival rate (71% at 5 years) when compared to patients who received palliative treatment [26]. Some others authors have also previously proposed, due to slow growth rate of the majority of malignant enteropancreatic tumors, surgery with cyto-reductive intent in patients with normal liver function despite gross tumor load and less than 90% of resectable tumor tissue [27], improving symptoms and quality of life however no evidence of prolonged survival in such cases. As for lymph node dissection, although lymph node involvement is a very common finding with malignant insulinoma, further studies are needed to assess the role of lymph node dissection in improving survival and preventing recurrence.
Moreover, patients with metastases at diagnosis and palliative treatment (54 of 64 cases reported) have a limited survival with an average period of two years [3,4]. In these advanced metastatic cases, a final resort combination chemotherapy using doxorubicin (fluorouracil if doxorubicin is contraindicated) and streptozotocin can be considered.
In our case, total pancreatectomy was achieved due to the barely remaining tissue of body and tail along with duodenectomy (classical, non-pylorus preserving), cholecystectomy, extensive lymph node dissection and re-establishment of the alimentary tract with roux en Y gastro-jejunostomy and hepatico-jejunostomy; achieving by this a complete excision of the insulinoma
Conclusion
The natural history of malignant insulinoma depends on the extension of the disease, the associated comorbidities and biological behavior of the tumor. Malignant insulinoma tends to be fairly indolent and slow growing of an average of 1.5 years from onset of symptoms to diagnosis as depicted earlier, however it does carry a poor unfavorable prognosis, as we mentioned earlier, that is mostly owed to presenting as an advanced, metastatic and unresectable tumor at time of diagnosis. On the contrary, our case brings into light a 30 years history of symptomatic hypoglycemia and clinically evident insulinoma that manifested as a malignant MEN1 insulinoma with lymph node metastasis that was successfully completely resected with outstanding post-operative recovery and resolution of the long standing debilitating hypoglycemia.
Declarations
Acknowledgements: We would like to acknowledge the efforts of the general surgery department, endocrinology and radiology department at the Saint George Hospital University Medical Center Beirut Lebanon.
Conflict of interests: The authors report no conflicts of interest.
Inform consent form: An inform consent was signed by the patient, authorizing access on his medical records and completion of this work
References
- Wilder RM, Allan FN, Power M et al. Carcinoma of the islands of the pancreas: hyperinsulinism and hypoglycemia. J Am Med Assoc. 1927; 89: 348–355.
- Jensen RT, Cadiot G, Brandi ML, de Herder WW, Kaltsas G, Komminoth P, Scoazec JY, Salazar R, Sauvanet A, Kianmanesh R; Barcelona Consensus Conference participants. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: Functional pancreatic endocrine tumor syndromes. Neuroendocrinology. 2012; 95: 98–119.
- Service FJ, Mcmahon MM, O’Brien PC et al. Functioning insulinoma: incidence, recurrence, and long-term survival of patients: A 60-year study. In: Mayo Clinic Proceedings, Elsevier, Amsterdam. 1991; 711–719.
- Hirshberg B, Cochran C, Kleiner, et al. Malignant insulinoma, Spectrum of unusual clinical features: A review of the literature. American Cancer Society. 2005; 104: 264–272.
- Crippa S, Zerbi A, Boninsegna L et al. Surgical management of insulinomas: short and long-term outcomes after enucleations and pancreatic resections. Arch Surg. 2012; 147: 261–266.
- Trup D, Farren B, et al. Clinical studies of multiple endocrine neoplasia type1. QJ Med. 1996; 89: 653–669.
- Vanderveen K, Grant C. Insulinoma. Cancer Treat Res. 2010; 153: 235-52.
- Jonkers YM, Claessen SM, Veltman JA, Geurts van Kessel A, Dinjens WN, et al. Molecular parameters associated with insulinoma progression: chromosomal instability versus p53 and CK19 status. Cytogenet Genome Res. 2006; 115: 289-97.
- Pape UF, Berndt U, Müller-Nordhorn J, Böhmig M, Roll S, Koch M, et al. Prognostic factors of long-term outcome in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer. 2008; 15: 1083-97.
- Pape UF, Jann H, Müller-Nordhorn J, Bockelbrink A, Berndt U, et al. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer. 2008; 113: 256-65.
- De Herder WW, van Schaik E, Kwekkeboom D, Feelders RA. New therapeutic options for metastatic malignant insulinomas. Clin Endocrinol (Oxf). 2011; 75: 277-84.
- Zonera Ashraf Ali. Insulinoma: A review of the literature. Lankenau Cancer Center. 2006; 24: 40–54.13.
- Falconi M, Eriksson B, Kaltsas G, Bartsch DK, Capdevila J, et al. Vienna Consensus Conference participants. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology. 2016; 103: 153–171
- Nikfarjam M, Warshaw AL, Axelrod L et al. Improved contemporary surgical management of insulinomas: A 25-year experience at the Massachusetts General Hospital. Ann Surg. 2008; 247: 165.
- Boukhman MP, Karam JH, Shaver J et al. Insulinoma experience from 1950 to 1995. West J Med. 1998; 169: 98
- Chambers AJ, Pasieka JL. Observation versus surgery for nonlocalized insulinoma. Difficult decisions in endocrine surgery. Springer, Berlin. 2018; 459–470.
- Hypoglycemic disorders. Service FJ. N Engl J Med. 1995; 332: 1144–1152.
- Gardner DG. Multiple endocrine neoplasia in Basic and clinical endocrinology. 6th ed. McGraw-Hill. 2001; 5322–5330.
- Gagel RF, Marx SJ. Multiple endocrine neoplasia in Williams Textbook of Endocrinology. 11th. Saunders. 2008; 1706–1719.
- Okabayashi T, Shima Y, Sumiyoshi T, et al. Diagnosis and management of insulinoma. World J Gastroenterol. 2013; 19: 829–837
- Sotoudehmanesh R, Hedayat A, Shirazian N, et al. Endoscopic ultrasonography (EUS) in the localization of insulinoma. Endocrine. 2007; 31: 238–241.
- Grover AC, Skarulis M, Alexander HR, et al. A prospective evaluation of laparoscopic exploration with intraoperative ultrasound as a technique for localizing sporadic insulinomas. Surgery. 2005; 138: 1003–1008.
- Kaltsas GA, Besser GM, Grossman AB. The diagnosis and management of advanced neuroendocrine tumors. Endocrine Reviews. 2004; 25: 458–511
- Hellman P, Lundstrom T, Ohrvall U, Eriksson B. Effect of surgery on the outcome of midgut carcinoid diseade with lymph node and liver metastases. World J Surg. 2002; 25: 991–997.
- . Chen H, Hardacre JM, Uzar A, Cameron JL, Choti MA. Isolated liver metastases from neuroendocrines tumors does resection prolong survival?. J Am Coll Surg. 1998; 187: 88–92.
- . Lo CY, et al. Pancreatic Insulinomas: A 15-year experience. Arch Surg. 1997; 132: 926–930.
- Chamberlain RS, Canes D, Brown KT, Saltz L, Jarnagia W, et al. Hepatic neuroendocrine metastases: does intervention alter outcomes? J Am Coll Surg. 2000; 190: 432–445.