
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
A case of benign recurrent intrahepatic cholestasis unlinked to ATP8B1 and ABCB11
Hany Eskarous1*; Anuragh Gudur2
1 Section of Gastroenterology and Hepatology, Temple University Hospital, USA.
2 Department of Medicine, University of Virginia Medical Center, USA.
*Corresponding Author: Hany Eskarous
Temple University Hospital, 3401 N Broad St,
Philadelphia, PA 19140, USA.
Email: hany.eskarous@tuhs.temple.edu
Received : Oct 25, 2021
Accepted : Dec 08, 2021
Published : Dec 15, 2021
Archived : www.jcimcr.org
Copyright : © Eskarous H (2021).
Abstract
Benign Recurrent Intrahepatic Cholestasis (BRIC) is a rare genetic disorder characterized by recurring episodes of jaundice. Two subtypes of BRIC have been well-characterized in the literature: BRIC I, caused by mutations in ATP8B1 gene, and BRIC II, triggered by mutations in ABCB11 gene. There are exceedingly rare cases of BRIC in which individuals do not have mutations in either of the associated genes, suggesting the possibility additional loci implicated in this disorder. Herein, we present a case of BRIC in a 21-year-old male who demonstrated clinical, biochemical and histological evidence of disease but lacked both of the associated mutations in ATP8B1 and ABCB11.
Citation: Eskarous H, Gudur A. A case of benign recurrent intrahepatic cholestasis unlinked to ATP8B1 and ABCB11. J Clin Images Med Case Rep. 2021; 2(6): 1481.
Introduction
Benign Recurrent Intrahepatic Cholestasis (BRIC) is a rare genetic disorder characterized by recurring episodes of jaundice due to poor biliary flow in hepatocytes. The disease typically manifests in an autosomal recessive inheritance pattern, with average age of onset ranging from infancy to young adulthood [1]. During episodes of cholestasis, patients present with intense pruritis and jaundice that may last up to several weeks [2]. Other symptoms include nausea, vomiting, poor appetite, and steatorrhea from fat malabsorption. Laboratory evaluation usually reveals conjugated hyperbilirubinemia, elevated Alkaline Phosphatase (ALP), and elevated ALT/AST. Two subtypes of BRIC have been well-characterized in the literature: BRIC I, caused by mutation in ATP8B1 gene, and BRIC II, triggered by mutation in ABCB11 gene [3-4]. Both genes encode for important hepatocyte proteins involved in the flow of bile. Liver biopsies during symptomatic episodes commonly demonstrate noninflammatory hepatocanalicular cholestasis without fibrosis. In periods of remission, liver histology is normal [5]. In clinical settings, diagnosis often requires a high degree of suspicion and is confirmed by genetic testing for ATP8B1 and/or ABCB11 mutations. However, there are exceedingly rare cases of BRIC in which individuals do not have mutations in either of the associated genes. Here in, we present a case of BRIC in a 21-year-old male who demonstrated clinical, biochemical and histological evidence of disease but lacked both of the associated mutations in ATP8B1 and ABCB11.
Case presentation
A 17-year-old male presented with a chief complaint of yellowing of his skin and eyes for the past year. In this time frame, he also reported pruritus, light colored stools, and dark brown urine. He also endorsed intermittent episodes of sharp, periumbilical abdominal pain that may last for a few minutes. His symptoms were not triggered by illness or stress. He denies using any new medications, supplements, or vitamins. He noted bruising on legs, but no other new changes in his skin. He denied fatigue, oral ulcers, fevers, chills, light headedness, dizziness, nausea, vomiting, diarrhea, constipation, joint paints, or urinary symptoms. There is no history of any melena, hematochezia, or hematemesis.
The patient lived at home with his parents, who urged him to seek medical attention. He worked as a dancer. He denied tattoos and has never had a blood transfusion. He had no known medication allergies. He denied alcohol use, smoking, and recreational drug use.
On exam, the patient had normal vital signs. He was visibly icteric. Abdomen was soft, non-tender, non-distended, and without hepatomegaly. Lab results revealed Alkaline Phosphatase of 370, ALT 96, AST 66, and conjugated hyperbilirubinemia. An extensive workup for transaminitis was unrevealing, including infectious hepatitis, alpha-1 antitrypsin disease, Wilson’s disease, hemochromatosis, autoimmune hepatitis, celiac disease, PBC, STDs, and HIV. Imaging with abdominal ultrasound demonstrated increased echogenicity of the liver reflecting steatosis, without biliary dilation.
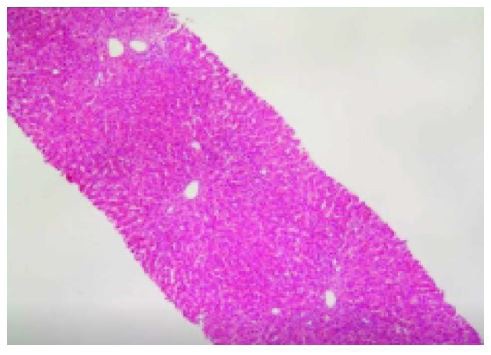
He also had negative genetic testing for the ATP8B1 and ABCB11 genes implicated in BRIC. A subsequent liver biopsy revealed bland cholestasis with coarse granular biliary material within dilated bile canaliculi, without any significant inflammation, bile duct damage, or ductular reaction (Figure 1). He was prescribed ursodiol plus hydroxyzine and discharged after reporting improvement in symptoms. Unfortunately, he discontinued those medications after discharge. He continued to experience intermittent episodes of jaundice and pruritis and ultimately presented to the hospital again with elevated Alkaline phosphatase, ALT, AST, and conjugated hyperbilirubinemia. A repeat workup for transaminitis, as completed previously, was unrevealing. He received ursodiol again and the symptoms resolved.
Discussion
We report a case of BRIC in the absence of mutations in ATP8B1 and ABCB11. Such cases are exceedingly rare in the literature. Initial cases of BRIC were first described in 1959, and a set of diagnostic criteria established in 1969 are still applicable today [6,7]. The criteria include: (1) multiple episodes of jaundice separated by a symptom-free interval of at least 6 months, (2) laboratory evidence of intrahepatic cholestasis, (3) histological evidence of noninflammatory cholestasis, (4) non-dilated biliary ducts, and (5) absence of other known risk factors for cholestasis.
Our patient satisfied these diagnostic criteria. The age of onset and history of symptoms align closely with the presentation of BRIC. Laboratory evaluation demonstrated a cholestatic pattern of liver enzyme elevation and conjugated hyperbilirubinemia, while abdominal ultrasound exhibited no biliary dilation-consistent with intrahepatic cholestasis. The pathologist’s assessment of the liver biopsy stated that the histopathology was compatible with a disorder of intrahepatic cholestasis, with morphologically similarities to biopsies seen in BRIC. Additionally, our patient lacked any other inciting factors to explain his clinical presentation.
Of note, some evidence indicates the existence of additional disease loci for low γ-GT BRIC [3,8,9]. This may explain why some individuals diagnosed with BRIC on clinical and histopathologic evidence do not have an association with either ATP8B1 or ABCB11. This disorder has been informally termed “BRIC 3”; it is unclear which genes are implicated in this form and whether they are inherited in an autosomal dominant or recessive pattern [10]. This case raises additional queries regarding the etiology of BRIC, which should prompt further investigation to characterize these additional genetic mutations that may be associated with the disease.
References
- Sticova E, Jirsa M, Pawłowska J. New Insights in Genetic Cholestasis: From Molecular Mechanisms to Clinical Implications. Can J Gastroenterol Hepatol. 2018; 2018: 2313675.
- Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. 2004; 8:133-149.
- Knisely AS, Bull LN, Shneider BL. ATP8B1 Deficiency. 2001. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. 1993-2021.
- Van Mil SW, Van der Woerd WL, Van der Brugge G, Sturm E, Jansen PL, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004; 127: 379-384.
- Biempica L, Gutstein S, Arias IM. Morphological and biochemical studies of benign recurrent cholestasis. Gastroenterology. 1967; 52: 521-535.
- Summerskill WH, Walshe JM. Benign recurrent intrahepatic obstructive jaundice. Lancet.1959; 2: 686-690.
- Velimir A. Luketic, Mitchell L. Shiffman, Benign Recurrent Intrahepatic Cholestasis, Clinics in Liver Disease, 1999; 3: 509-528.
- Floreani A, Molaro M, Mottes M, Sangalli A, Baragiotta A, et al. Autosomal dominant Benign Recurrent Intrahepatic Cholestasis (BRIC) unlinked to 18q21 and 2q24. Am J Med Genet. 2000; 95: 450–453.
- Strautnieks S, Byrne J, Knisely A, Bull LN, Sokal E, Lacaile F, Vergani G, Thompson R. There must be a third locus for low GGT PFIC. Hepatology. 2001; 34: 240A.
- Victoria EH Carlton, Ludmila Pawlikowska & Laura N Bull Molecular basis of intrahepatic cholestasis, Annals of Medicine. 2004; 36: 606-617.