
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Prenatal evaluation for the diagnosis of X-Linked myotubular myopathy in a known affected sibling: A case report
Vandana Bansal1; Meera Jayaprakash2*
1 Associate Professor, Department of Obstetrics and Gynaecology, Nowrosjee Wadia Maternity Hospital, Mumbai, India.
2 Fellow, Department of Obstetrics and Gynaecology, Nowrosjee Wadia Maternity Hospital, Mumbai, India.
*Corresponding Author: Vandana Bansal
Associate Professor, Department of Obstetrics and Gynaecology, Nowrosjee Wadia Maternity Hospital, Mumbai,
Maharashtra, India – 400012.
Email: drvandana_bansal@yahoo.co.in
Received : Nov 08, 2021
Accepted : Dec 29, 2021
Published : Jan 05, 2022
Archived : www.jcimcr.org
Copyright : © Bansal V (2022).
Abstract
X-linked myotubular myopathy, a rare disorder is part of a group of congenital myopathies that present in the neonatal period. While the clinical presentation in the antenatal period may be varied, the postnatal presentation is of a delayed cry, hypotonia and weakness resembling fetal hypoxia and acidosis. The presence of such features without any factors predisposing towards hypoxia, or a recurring phenotype should prompt a detailed evaluation of the neonate, including genetic evaluation in selected cases. A genetic diagnosis will also aid in evaluation of family members, including an early prenatal diagnosis in future siblings.
Keywords: X-linked myotubular myopathy; MTM1; prenatal diagnosis.
Abbreviations: X-MTM: X-Linked Myotubular Myopathy.
Citation: Bansal V, Jayaprakash M. Prenatal evaluation for the diagnosis of X-linked myotubular myopathy in a known affected Sibling: A case report. J Clin Images Med Case Rep. 2022; 3(1): 1537.
Introduction
Congenital myopathies are a rare group of phenotypically and genotypically diverse neuromuscular disorders that usually present in the neonatal period with respiratory distress, hypotonia and muscle weakness. They are defined on the basis of characteristic pathologic findings within muscle [1]. There are at least 32 different genetic causes of this disease, with a broad spectrum of histopathologic findings. However, many gene mutations may also share similar histopathologic findings [2]. Of these, the Myotubular or centronuclear myopathy is characterized by abundance of centrally located nuclei typically in the majority of type 1 fibres which are small in diameter; however, this can occur in both fibre types [3]. This may be transmitted in X-linked (MTM1 gene, Xq28), Autosomal Dominant (MYF6 gene, 12q21) and Autosomal recessive. (BIN1 gene, 2q14 [4]; TTN gene, chromosome 2 [5])
While the autosomal dominant form is slowly progressive and the autosomal recessive form is intermediate in severity and prognosis, the X-Linked Myotubular Myopathy (X-MTM) is a disorder that usually presents in infancy with prominent hypotonia and poor prognosis [3]. It is a rare condition (estimated in 1:50000 male births) associated with substantial morbidities and early mortality [6]. At present, being a rare disease with no current established treatments or disease-modifying therapies, care is based on generalized practice guidelines established for all congenital myopathies [7].
The clinical features of X-MTM have a broad spectrum of phenotypes. Severe (classic) X-MTM may be suspected in the antenatal period by polyhydramnios and decreased fetal movement. Neonatal weakness, hypotonia, respiratory failure being the norm, most neonates require 24-hour ventilator support. There is significant diffuse muscle weakness, diminished muscle bulk, cryptorchidism and involvement of the extra-ocular muscles (opthalmoparesis). However, the length and head circumference are greater than the 90th centile with long fingers and toes. Those with the severe phenotype rarely live into adulthood with 25% mortality in the first year of life. Males with mild to moderate X-MTM (~20%) achieve motor milestones relatively faster than those with the severe form, may ambulate independently and live till adulthood. However, many may still require ventilatory support and alimentation by gastrostomy [6]. There are case reports of males with delayed onset of the disease in adulthood [9]. Heterozygous females are usually asymptomatic, but symptomatic heterozygote females have also been reported with severity resembling affected males [9].
As the name suggests, X-MTM is inherited in an X-linked manner. If the disease is identified in a proband, there is 80- 90% probability that the mother of the proband is a carrier. If the mother is a carrier, the chance of transmitting the MTM1 pathogenic variant to her offspring is 50%. However, in 10-20% of cases, the mother is not found to be a carrier. This may represent de novo mutations or germline mosaicism. Carrier testing of at-risk female relatives and prenatal testing for pregnancies at-risk are possible if the pathogenic variant has been identified in a relative [8].
Case report
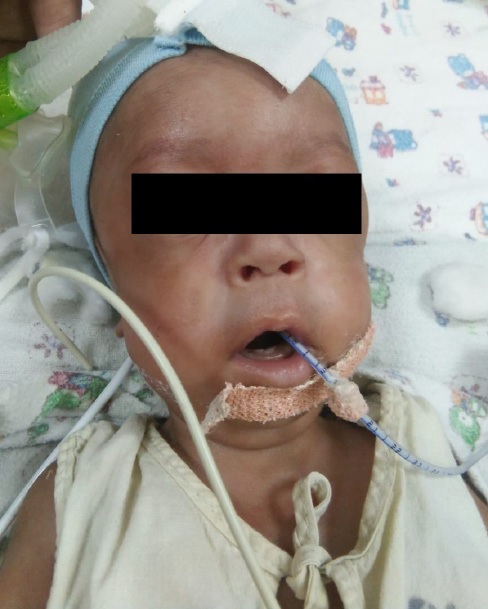
An apparently asymptomatic non-consanguineous couple presented to the Outpatient Department of a tertiary care hospital in Mumbai. The 28-year-old antenatal woman was a third gravida at 12 weeks of gestation. The first child, a male, was delivered vaginally at term with a birth weight of 1.9 kg. The delivery was relatively uneventful, however the baby had a delayed cry and died within a few hours of birth, as per the history given by the mother. No documents were available and the cause of death was not evaluated. It was assumed that the child died of birth asphyxia. In order to prevent a recurrence, her second child, also male was delivered by elective caesarean section. In spite of having an elective caesarean and in the absence of fetal distress or acidosis, this baby did not cry at birth. With a neonatal team already present, the baby was immediately intubated and shifted to ventilator in intensive care settings. Further neonatal evaluation revealed an absence of biochemical features of hypoxia or acidosis. Multiple attempts were made subsequently to wean the baby off the ventilator over the next few days. However, the baby could not be extubated even after multiple attempts due to a poor respiratory drive. In addition, the baby had an absent gag reflex, generalised hypotonia and areflexia and died in the sixth month of postnatal life.
The couple could not recall any history of similar disorder or unexplained neonatal or infant demise in their respective families. A clinical exome of the baby was advised, which detected a known pathogenic variant resulting in the reported phenotype.
The patient was counselled regarding the same and was advised to undergo genetic evaluation in her subsequent pregnancies.
In the present pregnancy, the patient presented at 12 weeks of gestation; hence it was decided to test the fetus of the patient for the same gene by chorionic villus sampling. The current fetus tested negative for the same mutation after ruling out maternal cell contamination, thus reassuring the prospective parents.
The mother was also offered testing for the same gene in order to direct genetic counselling for subsequent pregnancies. However, she wished to defer the same due to financial constraints.


Discussion
X-linked myotubular myopathy is a rare syndrome caused by mutations in the gene MTM1 which is located on the proximal segment of the long arm of the X chromosome. The characteristic clinical features are severe with muscle hypotonia and respiratory failure. Mortality is high in severe disease phenotypes; however milder variants have also been described. Similar features have been described in other centronuclear myopathies, which are inherited in an autosomal dominant or recessive pattern.
The variation in clinical phenotypes can be explained by the differences in the mutations. Milder disease variants had missense mutations whereas nonsense mutations resulting in a premature stop codon were found in most of the cases with the severe forms of the disease [1]. However, certain missense mutations may also cause the severe phenotype [2]. The index case was diagnosed with a nonsense variation that resulted in the truncation of the protein at codon 198, which has been described as a pathogenic mutation [3].
The index case was the second child of the couple. The previous child of the couple was also male, born with a low birth weight, had a weak cry at birth and died within a few hours. In the absence of a reasonable genetic suspicion and due to a low birth weight, the cause of death was probably ascribed to hypoxia. Hence the index case was delivered by elective caesarean section at term. In this case, the baby did not cry at birth and was immediately intubated and ventilated by a neonatal team that was in attendance during the elective caesarean. However, extubation of this neonate was not possible due to poor respiratory drive and hypotonia. The two apparently similar cases in the family prompted investigation of the index case.
The basic disorder was a poor respiratory drive and hypotonia, resulting in inability of the neonate to breathe. Similar clinical phenotypes can be found in many disorders such as congenital myopathies including centronuclear myopathies, congenital myasthenic syndromes and myotonic dystrophies [4,5]. Due to the genetic heterogeneity and a possible recurring incidence in consecutive pregnancies it was decided to evaluate the index case with a clinical exome sequencing. A clinical exome allows for the sequencing of disease-causing genes. At a fraction of the cost of whole exome sequencing (which covers the whole coding region of the genome), it allows for a relatively rapid reporting by next generation sequencing techniques [6].
A pathogenic variant was found in the clinical exome, which was correlating well with the clinical phenotype. The gene, located on the X-chromosome had a nonsense mutation that led to the truncation of the myotubularin protein, thus rendering it practically ineffective. The disorder is transmitted in an Xlinked fashion, which meant that 50% of the male offspring of the couple would be affected and that the mother would be a carrier for the same. However, there are reports in literature of heterozygous females who may show features of the disease which may be explained by X-chromosome inactivation or with more plausibility, an X-linked dominant inheritance pattern of the disorder with an incomplete penetrance among females [7]. There are also reports of de novo as well as germ-line mutations in 10-20% of cases with normal maternal genes. Hence, a prenatal diagnosis should be recommended to a couple for all subsequent pregnancies if the pathogenic variant is diagnosed in any offspring, due to the serious and debilitating nature of the disorder in male offspring by default, and a possibility of disease manifestation in females. Maternal carrier testing should be offered and if detected, the same may be recommended to her siblings.
The couple presented to us at the 12th week of gestation, hence it was decided to test the fetus by Sanger sequencing on a sample obtained by chorionic villus sampling after ruling out maternal cell contamination. Chorionic villus sampling is the preferred method of obtaining genetic material for the evaluation of single gene disorders [8].
The pathogenic variant was not detected in the fetus and hence the prospective parents were reassured. It was recommended to test for the same mutation in the mother with a view to further study the nature of the mutation and to possibly extrapolate testing among family members.
Conclusions
Birth asphyxia or intrapartum asphyxia is a leading cause of neonatal demise worldwide. However, it is imprecise to ascribe all incidences of delayed or no cry at birth to intrapartum asphyxia. A thorough assessment of clinical signs, biochemical parameters, major malformations and in this case, genetic evaluation leads to the proper evaluation of the disorder and prevention of its recurrence in subsequent pregnancies. A recurrent phenotype in pregnancies, consecutive or otherwise, should especially prompt a thorough genetic evaluation of the index child and the parents.
X-linked Myotubular Myopathy is a rare disorder with debilitating effects in affected individuals. Carriers may also manifest the disease phenotype. It is clinically similar to numerous disorders. Hence in the absence of a clinical diagnosis, Clinical Exome of the index case is a valuable and relatively less expensive test to diagnose pathogenic genes which are known to cause a disorder. An appropriate and detailed evaluation is extremely helpful in establishing a diagnosis and for the management of the index case. It is also helpful for determining the inheritance pattern and for directing evaluation as well as for prognostication of family members.
It is recommended to offer an early prenatal diagnosis of all the subsequent offspring of the couple since 10-20% of the mutations in absence of a maternal mutation are de novo or germ-line mutations. The fetus should preferably be tested by Sanger sequencing of a sample obtained by chorionic villus sampling after ruling out maternal cell contamination. In an ideal scenario, the mother should be tested preconceptionally for the MTM1 gene mutation by Sanger sequencing. However, a negative report in the mother should not preclude fetal testing. A high index of suspicion and a multidisciplinary approach can help to ensure a relatively stress-free pregnancy and favourable neonatal outcome.
References
- Tubridy N, Fontaine B, Eymard B. Congenital myopathies and congenital muscular dystrophies. Curr Opin Neurol. 2001; 14: 575-582.
- Gonorazky HD, Bönnemann CG, Dowling JJ, The genetics of congenital myopathies, In: Handbook of Clinical Neurology, Elsevier. 2018; Vol. 148: 549-564.
- Anthony DC, Frosch MP, DeGirolami U, Peripheral nerve and skeletal muscle, In: Robbins and Cotran-Pathologic Basis of Disease, Elsevier, 7th Ed 2005, 1340(t), Eds. Kumar V, Abbas AK, Fausto N, ISBN 0-8089-2302-1
- Nicot AS, Toussaint A, Tosch V, Kretz C, Wallgren-Pettersson C, Iwarsson E, et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007; 39: 1134–1139.
- Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, De Chene ET, Swanson LC et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. 2013; 81: 1205-1214.
- Das S, Dowling J, Pierson CR, X-linked centronuclear myopathy. In: Gene Reviews, Eds. Adam MP, Ardinger HH, Pagon RA, Wallace SE. Seattle WA: University of Washington; 1993-2017.
- Wang CH, Dowling JJ, North K, Schroth MK, Sejersen T, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol. 2012; 27: 363-382.
- Hoffjan S, Thiels C, Vorgerd M, Neuen-Jacob E, Epplen JT, et al. Extreme phenotypic variability in a German family with Xlinked myotubular myopathy associated with E404K mutation in MTM1. Neuromuscul Disord. 2006; 16: 749–753.
- Biancalana V, Scheidecker S, Miguet M, Laquerrière A, Romero NB, et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: Delineating diagnostic clues. Acta Neuropathol. 2017; 134: 889–904.
- Laporte J, Biancalana V, Tanner SM, Kress W, Schneider V, Wallgren-Pettersson C, et al. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat. 2000; 15: 393-409.
- Mc Entagart M, Parsons G, Buj-Bello A, Biancalana V, Fenton I, Little M, et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord. 2002; 12: 939-946.
- Bachmann C, Jungbluth H, Muntoni F, Manzur AY, Zorzato F, et al. Cellular, biochemical and molecular changes in muscles from patients with X-linked myotubular myopathy due to MTM1 mutations. Hum Mol Genet. 2017; 26: 320-332.
- Gilbreath HR, Castro D, Iannaccone ST. Congenital myopathies and muscular dystrophies. Neurol Clin. 2014; 32: 689-viii.
- Colombo I, Scoto M, Manzur AY, Robb SA, Maggi L, et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurology. 2015; 84: 28-35.
- Miyatake S, Matsumoto N. Genetics: Clinical exome sequencing in neurology practice. Nat Rev Neurol. 2014; 10: 676-678.
- Souza LS, Almeida CF, Yamamoto GL, Pavanello RCM, GurgelGiannetti J, da Costa SS, et al. Manifesting carriers of X-linked myotubular myopathy: Genetic modifiers modulating the phenotype. Neurol Genet. 2020; 6: e513.
- Wieacker P, Steinhard J. The prenatal diagnosis of genetic diseases. Dtsch Arztebl Int. 2010; 107: 857-862.