
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Desminopathy with early cardiac involvement mimicking apical hypertrophic cardiomyopathy: Demonstration by cardiovascular magnetic resonance
Anahita Tavoosi; Sophie Chenier; Nina Ghosh; Mariana M Lamacie; Andrew M Crean*
Center for Cardiovascular MRI, University of Ottawa Heart Institute, Ottawa, Canada.
*Corresponding Author: Andrew M Crean
Center for Cardiovascular MRI, University of Ottawa Heart
Institute, Ottawa, Canada.
Email: acrean@ottawaheart.ca
Received : Nov 06, 2021
Accepted : Dec 30, 2021
Published : Jan 06, 2022
Archived : www.jcimcr.org
Copyright : © Crean AM (2022).
Abstract
We present a report of a patient with desmin-related skeletal myopathy who underwent Cardiac MRI (CMR) following episodes of palpitations. Despite a recent normal echocardiogram, the CMR study revealed mild but unequivocal apical hypertrophy. These appearances were indistinguishable from those seen with sarcomeric apical hypertrophic cardiomyopathy. This case adds desminopathy to the growing list of potential HCM phenocopies and should be considered in situations where skeletal muscular weakness has also been identified.
Citation: Tavoosi A, Chenier S, Ghosh N, Lamacie MM, Crean AM. Desminopathy with early cardiac involvement mimicking apical hypertrophic cardiomyopathy: Demonstration by cardiovascular magnetic resonance. J Clin Images Med Case Rep. 2022; 3(1): 1538.
Introduction
Myofibrillar Myopathy (MFM) is characterized by a group of phenotypically and genotypically heterogenous neuromuscular disorders. One of these disorders is desmin-related myopathy, which has a predominant autosomal dominant mode of inheritance and is caused by a mutation in the Desmin Gene (Des) [1-3]. Individuals with desminopathy develop cardiomyopathy, cardiac conduction disease, and skeletal myopathy, in 49%, 60%, and 74% of cases, respectively [1]. The skeletal myopathy usually presents with bilateral weakness involving distal leg muscles spreading proximally, leading eventually to wheelchairdependence [2,4]. The most common form of myocardial involvement is Dilated Cardiomyopathy (DCM) [1,5,6] followed by Restrictive (RCM) [7-9], Arrhythmogenic (ACM) [10,11] and finally Hypertrophic Cardiomyopathy (HCM) [12,13]. Historically, these cases were diagnosed by echocardiography; as a result, it is likely that mild cases were missed. In the current era, CMR is often used, can detect tissue changes earlier, and may predict the prognosis by identification of cardiac fibrosis [14-17]. We describe a case of desminopathy, detected by CMR, with a previously unreported apical hypertrophic phenotype.
Case presentation
A 39-year-old man presented with a 6-year history of slowly progressive symmetric leg weakness (distal greater than proximal), and mild distal upper extremity weakness. He had preserved sensation in the upper and lower extremities, but absent ankle reflexes. There were no upper motor neuron signs. From a cardiac perspective, he was in NYHA functional class I-II. He described intermittent palpitations both at rest and with activity. There was no history of syncope or heart failure symptoms.
The patient reached both gross and fine motor developmental milestones on time with no delay. As a child, he was able to keep up with his peers and he participated in sports such as swimming, basketball, and soccer. He was able to run and jump as a child. He did not require any gait aids or braces during childhood.
The patient’s mother, already deceased, had muscle weakness. However, she was never formally assessed. A maternal aunt and a maternal cousin are also affected. His maternal aunt underwent genetic testing, which demonstrated a pathogenic mutation in the Des gene. The patient has three sisters and one brother, all asymptomatic. He has three daughters, all of whom are healthy.
He underwent EMG exam in 2017, which reported denervation in his gastrocnemius muscles bilaterally. His Creatine Kinase (CK) in 2016 was 907 IU/L and in 2017 was 925 IU/L (Normal range is 30–250 IU/L). Genetic testing confirmed that he was a heterozygous carrier for the familial pathogenic Des mutation (DES c.1034T>c p. Leu345Pro).
Cardiac screening was performed with echocardiogram, Holter monitor, and Cardiovascular Magnetic Resonance (CMR). The echocardiography showed prolapse of the anterior leaflet of mitral valve as well as mild dilatation of the left atrium. His ejection fraction was 60%. There was mild mitral regurgitation and mild tricuspid regurgitation on Doppler. No arrhythmia was detected on ambulatory monitoring. The cardiac phenotype by echo was otherwise unremarkable.
CMR was undertaken on a 1.5T system (Siemens Avanto, Siemens Medical Systems, Germany). Steady state free precession cine imaging demonstrated LV diastolic wall thickness of 9.5 mm in the apical segments of anterior and septal wall (Figure 1). Global and regional wall motion was normal (LVEF 63%). Late Gadolinium Enhancement (LGE) imaging was performed with Gadolinium (Gd)-DTPA 0.15 mmol/kg (Magnevist, Bayer Schering Pharma, Berlin, Germany), using a 2D inversion recovery prepared turboflash sequence. Native and post contrast T1 maps were also acquired. These revealed focal abnormal signal in the thickened apical segments (Figure 1), indicative of myocardial fibrosis.
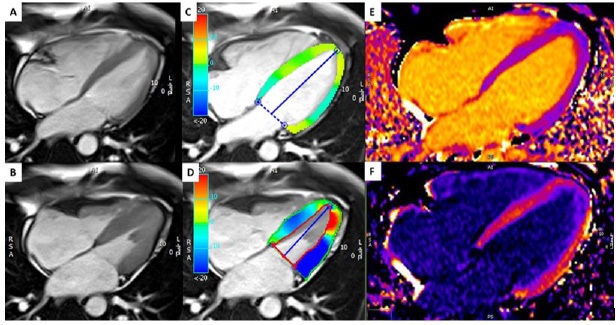
Discussion
Desmin-related myopathy, which is caused by accumulation of insoluble mutant desmin protein in skeletal, cardiac and striated muscle, has variable phenotypic expression [1]. Some patients will experience progressive skeletal myopathy without cardiac manifestations; others develop an isolated primary cardiomyopathy characterized as DCM or RCM. This differential phenotypic expression is due to the type and precise location of the mutation within the desmin gene [18,19]. In this case the presentation was a slowly progressive skeletal myopathy. The initial evaluation of cardiac involvement revealed no LV dilation or hypertrophy. CMR revealed increased myocardial thickness and LGE in the apical segments. The apical hypertrophy was not apparent at echocardiography and this is congruent with the experience of Moon et al who demonstrated that subtle apical HCM is frequently under-appreciated unless CMR is employed.
Strach et.al. evaluated eleven patients with pathogenic desmin mutations with both forms of imaging [14]. CMR revealed subtle focal hypertrophic myocardial changes in two patients (n=2/11), which were not detected by echocardiography. In addition, there were four patients (n=4/11) in whom LGE was detected at CMR, without corresponding global or regional wall motion abnormality at echocardiography.
He et al reported another case of desminopathy which presented as a LV thickened septal and lateral wall [13]. This finding in echocardiography was associated with LGE in the lateral wall detected by CMR. One further example of an HCM phenocopy, caused by a novel homozygous desmin mutation, did not go under cardiac MRI [12].
Conclusion
We present a case of cardiac involvement, in a patient with desminopathy, which would have gone unrecognized without CMR. Only knowledge of the patient’s history prevented this patient from being misdiagnosed as sarcomeric apical HCM. As far as we are aware this is the first case in the literature to describe a true apical phenotype in the context of a pathogenic desmin mutation.
References
- Ky VS, L VH, Jdh J, Walle D. Desmin-related myopathy. 2011; 354- 366.
- Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. Desmin myopathy. Brain. 2004; 127: 723-734.
- Kubánek M, Schimerová T, Piherová L, Brodehl A, Krebsová A, et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. J Clin Med. 2020; 9: 937.
- Goldfarb LG, Olivé M, Vicart P, Goebel HH. Intermediate filament diseases: Desminopathy. Adv Exp Med Biol. 2008; 642: 131-164.
- Li D, Tapscoft T, Gonzalez O, Burch PE, Quiñones MA, et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999; 100: 461-464.
- Taylor MRG, Slavov D, Ku L, Di Lenarda A, Sinagra G, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007; 115: 1244-1251.
- Koitka K, Dahiya A, Lo A, Scalia GM, Atherton JJ, Prasad SB. Myofibrillar Cardiomyopathy due to a Novel Desmin Gene Mutation: Complementary Role of Echocardiography, Cardiac Magnetic Resonance, and Genetic Testing in Delineating Diagnosis. Case. 2017; 1: 28-33.
- Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail. 2006; 8: 477-483.
- Olivé M, Armstrong J, Miralles F, Pou A, Fardeau M, et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord. 2007; 17: 443-450.
- Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010; 19: 4595-4607.
- Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IEA, et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013; 111: 400-405.
- Harada H, Hayashi T, Nishi H, Kusaba K, Koga Y, et al. Phenotypic expression of a novel desmin gene mutation: Hypertrophic cardiomyopathy followed by systemic myopathy. J Hum Genet. 2018; 63: 249-254.
- He Y, Zhang Z, Hong D, Dai Q, Jiang T. Myocardial fibrosis in desmin-related hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2010; 12: 2-4.
- Strach K, Sommer T, Grohé C, Meyer C, Fischer D, et al. Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. Neuromuscul Disord. 2008; 18: 475-482.
- Silva MC, Meira ZMA, Gurgel Giannetti J, da Silva MM, Oliveira Campos AF, et al. Myocardial Delayed Enhancement by Magnetic Resonance Imaging in Patients With Muscular Dystrophy. J Am Coll Cardiol. 2007; 49: 1874-1879.
- Suk T, Edwards C, Hart H, Christiansen JP. Myocardial Scar Detected by Contrast-Enhanced Cardiac Magnetic Resonance Imaging is Associated with Ventricular Tachycardia in Hypertrophic Cardiomyopathy Patients. Hear Lung Circ. 2008; 17: 370–374.
- Cardiac magnetic resonance detection of myocardial scarring in hypertrophic cardiomyopathy: Correlation with histopathology and prevalence of ventricular tachycardia-PubMed. 2021.
- Bär H, Mücke N, Kostareva A, Sjöberg G, Aebi U, et al. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc Natl Acad Sci U S A. 2005; 102: 15099-15104.
- Kostera Pruszczyk A, Pruszczyk P, Kamińska A, Lee HS, Goldfarb LG. Diversity of cardiomyopathy phenotypes caused by mutations in desmin. Int J Cardiol. 2008; 131: 146-147.