
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Pachygyria in a Saudi preteen female: A case report and review of literature
Oluniyi S Afolabi*; Ahmed Mohamed Ezzeldeen Mahmoud
Department of Radiology, King Fahad Hospital, Kingdom of Saudi Arabia.
*Corresponding Author: Oluniyi S Afolabi
Department of Radiology, King Fahad Hospital,
Kingdom of Saudi Arabia.
Email: afosemes@gmail.com &
oafolabi@moh.gov.sa
Received : Nov 15, 2021
Accepted : Jan 04, 2022
Published : Jan 11, 2022
Archived : www.jcimcr.org
Copyright : © Afolabi OS (2022).
Abstract
Pachygyria is a component of a spectrum of malformation of cerebral neuronal migration. MRI is a fundamental neuroimaging tool for adequate description of cerebral structural pathology. As a component of lissencephaly-pachygyria spectrum, there are severe intertwined clinical and pathological manifestations, with/without other brain and systemic malformations. Clinical features include protracted seizures, delay or absence of developmental milestones, and varying degrees of mental retardation.
This is a case of a preteen with previous infantile seizures and delay in developmental milestones. Her MRI showed uncommon but classic “leopord” and “tigroid” appearances of deep white matter changes, as well as the typical shallow and few cerebral cortical sulci, as well as cortical thickening of pachygyria. Despite several established genetic mutations documented in various disorders of cerebral neural migration, there was however no genetic assay yet for the patient at the time of this publication.
Keywords: pachygyria; lissencephaly; neuronal-migration; heterotopia.
Citation: Afolabi OS, Mahmoud AME. Pachygyria in a Saudi preteen female: A case report and review of literature. J Clin Images Med Case Rep. 2022; 3(1): 1547.
Introduction
Pachygyria is a neurodevelopmental anomaly in which there is abnormality of brain cortical neuronal cellmigration. It is a component of developmental abnormality called Lissencephaly-Pachygyria spectrum [1]. There are generally a few and shallow cortical sulci with or without band heterotopia [1]. Lissencephaly, basically signifies complete absence of sulci (agyria) giving the so-called smooth brain appearance, while in pachygyria, there are some few and shallow sulci. There is a strong association with band heterotopia in the Lissencephaly type I (Classic), in which there is usually a parallel grey matter tissue within the subcortical white matter, giving the so-called double cortex appearance [1]. Generally, lissencephaly type I is a rare disease condition with a prevalence of about 11.7/million and a female gender predominance in the Netherlands. There was a documentation of 5 year survival ranging between 54% and 91% [2]. Prevalence of lissencephaly in the United States is estimated to be 1.2/100000 births. [3]
Disorder of neuronal migration could be due to under-migration (Lissencephaly type I) or over-migration (Lissencephaly type II) neuronal cells. The Lissencephaly type II is also called the cobblestone lissencephaly, due to the pebble-like brain surface [4]. Several genetic aetiologies have been reported with various manifestation of brain cortical malformations. These could include PAFAH1B1 (LIS1), microdeletions of 17p13.3, tubulin encoding gene deletions (TUBA1A, TUBG1 and TUBGCP6), mutations in the DCX, DYNC1H1, ADGRG1, and WDR62 genes. Other genetic aetiology could include intragenic deletion in TRIO [5-7]. There had been a documentation of specific imaging features which correspond to certain genetic aetiologies [5]. The mutations of FLNA gene had been documented in all familial cases of X-linked periventricular nodular heterotopia, and in about one-fourth of sporadic cases [8].
Cross sectional radiological imaging plays a major role in the diagnosis of pachygyria and other brain cortical malformations. Even though ultrasound and computed tomography could play some roles in diagnosis, Magnetic Resonance Imaging (MRI) remains the imaging modality of choice. MRI is fundamentally responsible for comprehensive description and classification of the pathologies [1,9]. Pachygyria, apart from the fundamental few and shallow sulci, presents with varying degree of cerebral cortical thickening, abnormality of angulation of the sylvian fissure, basal ganglia anomalies, regional agyria or pachygyria in different part of the brain cortices and abnormalities of corpus callosum [5,10]. Clinical features may include seizures, delay in developmental mile-stones, mental retardation, hypotonia, facial dysmorphism, ambiguous genitalia (ARX gene), microcephaly, limb abnormality and gastrointestinal malformation [5,10-13].
Case report
This is case of a 12 year old Saudi Female who has been on follow-up management for developmental delay since age 2 years. There were seizures at infancy. Patient had malocclusion, for which she had tooth extraction at age 3 years. She also had visual and hearing partial losses. There was prolactinaemia.
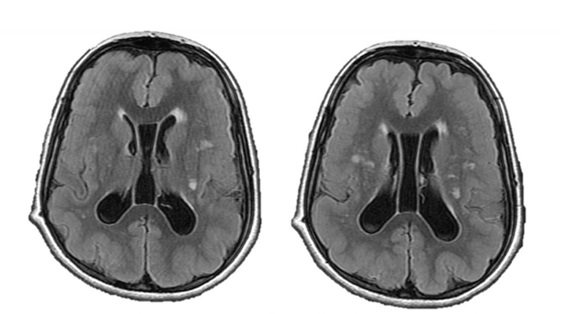
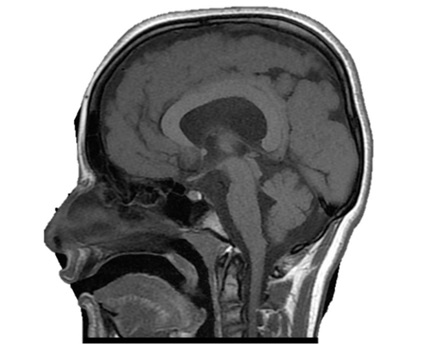
MRI images showed shallow and few cortical sulci and poorly formed gyri, with gross thickening of the brain cortex ranging between 16 mm and 21 mm. There was diffuse involvement of the fronto-temporo-parieto-occipital cortices. Diffuse periventricular white matter hyperintensities, as well as perpendicularly oriented deep white matter hyperintensities were noted. Patient had dilatation of the lateral ventricular system with persistence of the cavum septum pellucidum and cavum vergae. There was asubependymal heterotopia in the left lateral ventricle. Basal ganglia, brainstem, cerebellum and corpus callosum were however preserved (Figure 3).
Discussion
Developmental delay [14,15] and various forms of seizures [5,14-17] as demonstrated in this patient are known clinical presentations of Pachygyria. Several authors have reported myriads of related/overlapping clinical presentations in pachygyria and other disorders of neural migration, such as heterotopias. Di Nora A. et al studied the clinical and neuroimaging manifestations of various grey matter heterotopias in 22 patients in a tertiary institution in Italy. Periventricular Heterotopia (PVH) was seen reported in seventeen, Subcortical Band Heterotopia (SBH) in two and Subcortical Heterotopia (SUBH) in three of the cases. Developmental delay and intellectual deficits were the most predominant clinical presentations seen in over 54% of the patients, followed by various forms of epileptic seizures (mostly focal to bilateral tonic-clonic) in 11 (50%) of the cases. Of the 17 PVHs, twelve of them showed cerebral malformations, while system malformations were reported in ten. Three of the five children presented with SBH and SUBH, three presented with epileptic seizures, two manifested systemic malformations and only one showed cerebral malformations. There was also male predominance [18]. Similar findings were reported by Raza H. A. et al in China in which fifteen paediatric and adult patients with grey matter heterotopia were retrospectively analysed. There was a male predominance, as the authors reported thirteen cases with epilepsy, of which five showed cognitive decline and three showed neurological deficit. The remaining two cases only showed headache or dizziness. Seizures in these patients were generally poorly controlled, probably depicting the extent of severity of the pathologies. Grey matter heterotopias were reported, just like in this index case where there was left subependymal heterotopia. Raza et al however reported varying forms of heterotopias which were either unilateral, or multifocal in the periventricular, subependymal, subcortical and centrum semi ovale. These were further classified into two basic groups: the subcortical and subependymal/periventricular classes, respectively demonstrated by ten and five individuals. There were associated other brain pathologies in most of the cases. Other brain pathologies seen included ventricular compression in eight patients, ventricular dilatation/ enlargement in five patients, cerebral fissure malformation in five patients, pachygyria in three patients, and corpus callosal agenesis and hypoplastic septum pellucidum in one case each [10]. Ventricular dilatation, however without compression or callosal abnormality was noted in this Saudi female case of pachygyria (Figure 2 & 3). Another study from a Taiwanese tertiary health institution presented similar clinical and neuroimaging findings in thirty-six children with grey matter heterotopias. Unlike, the previously discussed studies, there was a slight female predominance. In this particular publication, the cases were divided into two fundamental classes, based on the location of the heterotopias. Namely: the periventricular heterotopias and band heterotopias. Twenty- six patients demonstrated the periventricular heterotopia, while the remaining ten showed band heterotopia in location. There were associated cerebral malformations in eighteen, and systemic malformations in sixteen of the periventricular heterotopia group. Similar pattern, but in lower percentage was also reported in the band heterotopia group, which respectively showed five and two cases with additional cerebral and systemic malformations. The predominant cerebral malformations included ventriculomegaly, corpus callosum abnormality and Dandy-Walker cyst, while congenital heart disease was commonest systemic malformation. Majority (twenty-two) of the overall cases showed various forms of seizures, of which more than half (twelve) showed refractory pattern [19]. Heterotopia (subependymal) and ventriculomegaly were similarly demonstrated in this index case, as seen in “Figure 2”. Some uncommon CNS and ocular pathologies, such as visual and hearing impairments, as well as malocclusion which were not reported in the reviewed literature.
The fundamental imaging presentations of Pachygyria which include few gyri and shallow sulci, as well as cortical thickening were all noted in this female patient. The demonstration of gross cortical thickening was also reported by Schaffer A. E. et al, although the comparative study showed higher level of tissue thickening, worse than in the index case (1.6-2.1 cm vs 3-4 cm) (Figure 2) [20]. A rare imaging pattern of leopard (sagittal/coronal) appearances of T2 and FLAIR respective perpendicularly/ radially oriented and dotted (axial) white matter hyperintensities in deep/ subcortical white matter reported by Roy U et al were also seen in the reported case. However, the leopord pattern was demonstrated in the axial and coronal images, while tigroid (dotted) pattern was seen in the sagittal images, in contrast to what was previously described by Roy U et al [21]. This is described in “Figure 1”. This imaging patterns were previously reported in metachromatic leukodystrophy and Pelizaeus-Merzbacher disease, and are fundamentally a type of demyelination [21].
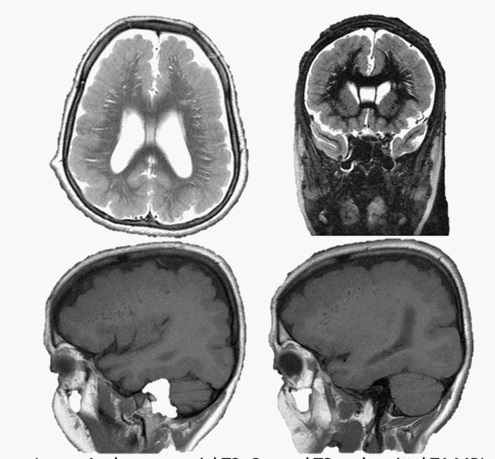
Pachygyria, being classified as a variant of Lissencephaly, has been documented to be associated severe genetic pathologies/ mutations. However, genetic assay had not been done, as at the time of this publication. Romero D et al was able to demonstrate genetic associations of each of the various manifestation of malformation of cortical migration, which may include Cobblestone lissencephaly, Periventricular Heterotopia (PVH), Subcortical Band Heterotopia (SBH), Agyria lissencephaly, Congenital microcephaly, and microcephaly with lissencephaly. For example, the cobblestone lissencephaly was shown to be associated with FKTN, POMT1, POMT2, POMGNT2, POMK, FKRP, LARGE, LAMB1, TMTC3, ISPD, TMEM5, B4GAT1, GPR56, B3GALNT2, and DAG1. Periventricular heterotopia was reported to be associated mutations of FLNA, ARFGEF2, C6orf70, FAT4, DCHS1, LRP2, and NEDD4L. LIS1, DCX, ARX, TUBA1A, TUBB3, TUBG1, CDK5, VLDLR, ACTG1, ACTB, and DYNC1H1, RELN gene abnormalities have respectively been associated with Agyria lissencephaly and Agyria lissencephaly with microcephaly [7]. Just like Romero D et al, Kolbjer S. et al also reported specific genetic mutations implicated in various forms of lissencephaly. Twenty patients with associated epilepsy, ages ranging between eighteen months and twenty-one years were analysed. There was male predilection in this particular study. Eleven individuals (55%) showed mutations in PAFAH1B1 (LIS1) gene or variable microdeletions of 17p13.3. About one-fifth (4 patients) of the cases demonstrated tubulin encoding gene (TUBA1A, TUBG1 and TUBGCP6) mutations. Only one of the patients shows mutations of the DCX, DYNC1H1, ADGRG1 and WDR62. Kolbjer S. et al also showed possible intragenic deletion in the TRIO gene. They reported distinct imaging presentations in patients with PAFAH1B1 and tubulin encoding genetic pathologies. Patients with the former (PAFAH1B1) showed diffuse agyria or posterior dominant pachygyria with significant cortical thickening, while in the latter (tubulopathies), the patients showed perisylvian pachygyria and preservation of normal thickness of the brain cortex. Only imaging-confirmed agyria was able to reliably predict molecular results with a posterior to anterior gradient [22]. Schaffer A. E. et al was able to demonstrate specific genetic mutation in three consanguineous families, which had seven Pachygyria individuals diagnosed before age two years. Majority (5/7) of the cases were from Saudi Arabia, which might represent higher incidence. The cases showed varying forms of developmental delays, including microcephaly, cerebral palsy, gross and fine motor delay or absence, speech defect and social development. They also presented with various forms of seizures, tone and deep tendon abnormalities. The imaging manifestations comprises MRI-confirmed pachygyria with thickened cortex up to 3-4 cm, absent anterior commissure, corpus callosum and cerebellar hypoplasia [20]. Schaffer A. E. et al was also able to affirm a common biallelic truncating mutations in CTNNA2, encodingαN- catenin in these patients who demonstrated recessive form of pachygyria. This aforementioned CTNNA2 gene is basically expressed in the human cerebral cortex, the defect of which leads to loss of neuronal stability and cortical migration. Schaffer and her colleagues were able to identify CTNNA2 as the first catenin family with biallelic mutations in human beings, with its resultant pachygyria syndrome [20].
To further emphasize the genetic aetiologies of pachygyria, an association with trisomy 21 (Down syndrome) was reported by Shabih Manzar. This was a male patient, a second of a set of dizygotic dichorionic foetuses, delivered via caesarean section at 28 weeks gestational age. In addition to the general phenotypic appearance of Down syndrome, the patient also showed MRI features of pachygyria, however without midline or callosal pathology [23].
Abdel Razek et al attempted to analyse the embryologic stages of the cerebral cortex, describe classification of the pathologies of cortical malformation and eventually illustrate the magnetic resonance imaging features of these brain malformations. The various cortical abnormalities are grouped based on the cerebral embryologic stage at which the insult occurred. Microcephaly was reported to demonstrate significant cortical thinning and loss of sulcation, hemimegaencephaly was reported to show enlarged dysplastic cortex, and focal cortical dysplasia stated to demonstrate ipsilateral focal cortical thickening with radial hyperintense bands. As reported in various studies, classic lissencephaly was stated to show smooth brain, congenital muscular dystrophy was reported to show nodular cortex with cobblestone cortex, and heterotopias show ectopic/ abnormal grey matter locations [9].
Conclusion
This a case of pachygyria, a variant of lissencephaly which is a rare malformation of cerebral neuronal migration, in which an uncommon pattern of white matter disease was demonstrated. Patient was however seen to show the usual clinical pattern of seizures, developmental delay and other central nervous impairment, as well as classic neuroimaging features.
Limitation: A genetic association could not be ascertained, as no genetic assay had been done as of time of this publication.
Declarations
Consent: A verbal consent was obtained from the parents, having assured them of maintaining strict patient’s anonymity in all writings and images involved in this publication.
Funding and declaration of competing interest: There was neither external funding nor any competing interest.
Acknowledgement: The authors acknowledge and appreciate Mr Jamaan and Mr Ahmed of the of the IT department of Radiology in making the images available/ downloadable for this publication.
References
- Lissencephaly-pachygyria spectrum. Radiology Reference Article. Radiopaedia.org [Internet]. [cited 2021 Oct 15]. Available from: https://radiopaedia.org/articles/lissencephaly-pachygyria-spectrum-2
- Andel JF de R, Arts WFM, Hofman A, Staal A, Niermeijer MF,et al. Epidemiology of Lissencephaly Type I. Neuroepidemiology. 1991; 10: 200–204.
- Lissencephaly NORD (National Organization for Rare Disorders) [Internet]. [cited 2021 Oct 12]. Available from: https://rarediseases.org/rare-diseases/lissencephaly/
- Lissencephaly type II. Radiology Reference Article. Radiopaedia. org [Internet]. [cited 2021 Oct 15]. Available from: https://radiopaedia.org/articles/lissencephaly-type-ii
- Kolbjer S, Martin D A, Pettersson M, Dahlin M, Anderlid B. Lissencephaly in an epilepsy cohort: Molecular, radiological and clinical aspects. European Journal of paediatric neurology. 2021; 30: 71-81.
- Bahi-Buisson N, Guerrini R. Diffuse malformations of cortical development. Handbook of Clinical Neurology. 2013; 111: 653– 665.
- Romero DM, Bahi-Buisson N, Francis F. Genetics and mechanisms leading to human cortical malformations. Seminars in Cell and Developmental Biology. 2018; 76: 33–75.
- Parrini E, Guerrini R. Lissencephaly Type I and Periventricular Heterotopia. Encyclopedia of Neuroscience. 2009; 503–512.
- Razek AAKA, Kandell AY, Elsorogy LG, Elmongy A, Basett AA. Disorders of cortical formation: MR imaging features. American Journal of Neuroradiology. 2009; 30: 4–11.
- Raza HK, Chen H, Chansysouphanthong T, Zhang Z, Hua F, Ye X, et al. The clinical and imaging features of gray matter heterotopia: A clinical analysis on 15 patients. Neurological Sciences. 2019; 40: 489–494.
- Dahnert W. Brain Disorders. In: Radiology Review Manual. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 303– 304.
- Neuronal Migration Disorders Information Page. National Institute of Neurological Disorders and Stroke [Internet]. [cited 2021 Oct 12]. Available from: https://www.ninds.nih.gov/Disorders/ All-Disorders/Neuronal-Migration-Disorders-Information-Page
- Pachygyria. Genetic and Rare Diseases Information Center (GARD) – an NCATS Program [Internet]. [cited 2021 Oct 12]. Available from: https://rarediseases.info.nih.gov/diseases/7300/pachygyria
- Matthew T, Srikanth S G, Satishchandra P. Malformations of Cortical Development (MCDs) and epilepsy: Experience from a tertiary care center in south India. Seizure. 2010; 19: 147-152.
- Chang J, Zhao L, Chen C, Peng Y, Xia Y, Tang G, et al. Pachygyria, seizures, hypotonia, and growth retardation in a patient with an atypical 1.33 Mb inherited microduplication at 22q11.23. Gene. 2015; 569: 46–50.
- Liang JS, Lee WT, Young C, Peng SS, Shen YZ. Agyria-pachygyria: Clinical, neuroimaging, and neurophysiologic correlations. Pediatric Neurology. 2002; 27: 171–176.
- Kurul S, Çakmakçi H, Dirik E. Agyria-pachygyria complex: MR findings and correlation with clinical features. Pediatric Neurology. 2004; 30: 16–23.
- Di Nora A, Costanza G, Pizzo F, Oliva CF, Di Mari A, Greco F, et al. Gray matter heterotopia: Clinical and neuroimaging report on 22 children. Acta Neurologica Belgica. 2021.
- Hung PC, Wang HS, Chou ML, Lin KL, Hsieh MY, Wong AMC. Clinical and neuroimaging findings in children with gray matter heterotopias: A single institution experience of 36 patients. European Journal of Paediatric Neurology. 2016; 20: 732–737.
- Schaffer AE, Breuss MW, Caglayan AO, Al-Sanaa N, Al-Abdulwahed HY, Kaymakçalan H, et al. Biallelic loss of human CTNNA2, encoding αN-catenin, leads to ARP2/3 complex overactivity and disordered cortical neuronal migration. Nature Genetics. 2018; 50: 1093–101.
- Roy U, Pandit A, Das U, Panwar A. “Reverse Tigroid” Pattern in Pachygyria: A Novel Finding. Journal of Clinical Imaging Science. 2016; 6: 1-5.
- Kolbjer S, Martin DA, Pettersson M, Dahlin M, Anderlid BM. Lissencephaly in an epilepsy cohort: Molecular, radiological and clinical aspects. European Journal of Paediatric Neurology. 2021; 30: 71–81.
- Shabih Manzar. Pachygyria in a neonate with trisomy 21. Ann Saudi Med. 2007; 27: 122–124.