
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Series - Open Access, Volume 3
Rare case series of congenital adrenal insufficiency in two brothers with NROB1 mutation, presenting with dichotomous pubertal presentation
Versha Rani Rai; Ikramullah Shaikh; Karishma Rahak*; Roshia Parveen; Jamal Raza; Taj Muhammad Laghari; Mohsina Noor Ibrahim
National Institute of Child Health, Karachi, Pakistan.
*Corresponding Author: Kirishma Rahak
National Institute of Child Health, Karachi, Pakistan.
Email: dr.rahak@yahoo.com
Received : Dec 23, 2021
Accepted : Jan 13, 2022
Published : Jan 20, 2022
Archived : www.jcimcr.org
Copyright : © Rahak K (2022).
Abstract
Congenital Adrenal Hypoplasia (AHC) first described in 1948, characterized by adrenal insufficiency, and hypo-gonadotropic hypogonadism. X linked AHC mainly affects males, it occur mainly due to mutation in NR0B1 gene. In this report, we present two cases of NR0B1 mutation from same family with dichotomous pubertal presentation. An infant diagnosed and being managed as case of adrenal insufficiency, developed precocious puberty at the age of 11 months. His work-up showed increased LH, FSH and Testosterone. Genetic Analysis revealed NR0B1 mutation. He was managed with Gn RH agonist and steroid replacement. His brother 15 years old, treated as primary adrenal insufficiency, presented with arrested puberty, his genetic analysis also showed NR0B1 mutation.
Keywords: AHC; adrenal hypoplasia; precocious puberty; hypogonadotropic; hypogonadism.
Citation: Rai RV, Shaikh I, Rahak K, Parveen R, Raza J. Rare case series of congenital adrenal insufficiency in two brothers with NROB1 mutation, presenting with dichotomous pubertal presentation. J Clin Images Med Case Rep. 2022; 3(1): 1592.
Introduction
Congenital Adrenal Hypoplasia secondary to NR0B1 mutation consists of both Xp21 deletion (previously called complex glycerol kinase deficiency) and X-linked adrenal hypoplasia congenital (X-linked AHC). DAX1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1) is nuclear receptor protein formed in body under instruction of NR0B1 gene. NR0B1 (nuclear receptor, sub family 0 group B, member 1), located on NR0B1 gene on short (p) arm of X chromosome between bands Xp21.3 and Xp21.2, from base pair 30,082,120 to base pair 30,087,136. NR0B1 gene encodes protein which lacks usual DNA-binding domain present in other nuclear receptors. This protein acts as a dominant negative regulator of transcription of other nuclear receptors including steriodogenic factor 1 and also function as an anti-testis gene by acting antagonistically to SRY. Transcription factors are proteins which control activity of other genes. DAX1 protein plays pivotal part in the development and function of different endocrine glands. These includes adrenal glands, hypothalamus, pituitary, ovaries and testes. During fetal life, DAX1 protein helps regulate genes that instruct the formation of these gonads and following there formation also helps in regulation of hormone production in these glands.
Case series
Case 1
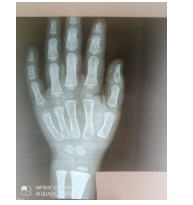
A neonate on 15th day of life presented with adrenal crises and dark pigmentation, he is 7th product of consanguineous marriage with history of two abortions and one sibling death at the age of one month, he was diagnosed as primary adrenal insufficiency and being managed with hydrocortisone and florinef. At the age of 9 month he was hospitalized due to COVID-19 pneumonia, later recovered and discharged to home. He remains well on the follow up, serial growth monitoring showed adequate weight gain and decreased in pigmentation. At the age of 11 month he presented with penile enlargement and appearance of pubic hairs with no history of drug exposure (androgens), fit, trauma or visual disturbance. At the time of examination, he was vitally stable with weight and height lies at -0.3 SDS. There was neither underarm hair growth nor neurocutaneous markers. Tanner Staging was P3G3 with testicular volume of 6 ml/6 ml. His penile length was 5 cm. Hormonal profile at the age of 11 month showed increased FSH, LH and Testosterone, while 17-OH-progesterone, Renin, ACTH and short synacthen test were at normal (Table 1). X-ray left wrist showed bone age of 3 years according to Greulich and Pyle’s atlas method (Figure 1). U/S Abdomen and inguinal region showed normal adrenal glands, enlarged testes with normal texture approximately measuring 3.8 X 1.8 X 1, 8 with volume of 6 ml. Genetic mutation revealed NROB1 mutation hemizygous (variant c.327C>A (p.cys109). He was treated with 5 monthly injections of Gn RH agonist and steroid replacement therapy. At the age of 16 months he was started with Injlutrate I/M. After 6 month on treatment his testicular volume remained unchanged where as pubic hair slightly reduced with Tanner staging P2 G2. Serial testosterone monitoring showed decreasing levels (Table 2).
Table 1:Hormonal Profile at the age of 11 months
DATE |
24/07/20 |
Reference range |
17-OH PROGESTERONE (ng/ML) |
1.0 |
0.03-0.9 |
RENIN (uIU/ml) |
49 |
2.8-39.9 (S) |
|
4.4-46.1 (E) |
|
LH (mIU/ml) |
1.41 |
( 0.8-1.3) |
FSH (mIU/ml) |
1.0 |
(0.5-2.4) |
TESTOSTERONE (ng/dl) |
472.4 |
(<30ng/dl) prepubertal |
CORTISOL (ug/dl) |
7.3 |
5-15 |
ACTH (pg/ml) |
123 |
10-50 |
Table 2:Serial monitoring of Testosterone.
11th month of life |
14th month of life |
17th month of life |
|
Testosterone |
472.4 |
246 |
122 |
Case 2
His brother 15 years old, presented with adrenal crises for 1 month and treated as primary adrenal insufficiency. At the age of 15 years, he presented with arrested puberty with Tanner scoring III and bilateral testicular volume 6/6 for past 1 year. His genetic work-up showed same NR0B1 hemizygous mutation.
Discussion
Hypotrophy or under development of adrenal cortex is known as Adrenal hypoplasia. It is divided into two categories, primary and secondary Adrenal hypoplasia. In primary Adrenal hypoplasia patients presented during infancy & childhood. It is either due to adrenal hypoplasia congenita or intrinsic defect in adrenal gland during development [1,2].
There are 4 forms of AHC describes in literature;
1- X-linked form due to mutation or deletion of DAX-1 gene
located on X-chromosome
2- Autosomal recessive form, whose gene located on chromosome (9q33) which encode steroidogenic factor 1 (SF1)
3- Rare Autosomal form such as IMAGE syndrome, SERKAL
syndrome
4- Autosomal recessive ACTH resistance type syndrome,
such as triple-A syndrome [1].
X-linked AHC, due to mutation in DAX-1 gene or contiguous gene deletion syndrome in association with Glycerol Kinase (GK) deficiency, Duchenne muscular dystrophy and X-Linked Interleukin-1 Receptor Accessory Protein-Like 1 (IL1RAPL1) gene deficiency [1,2].
As discussed above, DAX-1 protein plays very important role in in the development of the adrenals and hypothalamic-pituitary-gonadal axis. X-linked AHC presented as Adrenal insufficiency and/or Hypogonadotrophic Hypogonadism (HH). About 40% of affected males presents with adrenal insufficiency during infantile age (average 3 weeks) and rest of 40% present during childhood (age 1-9 years) [3,4].
In X-linked AHC, Hypogonadotrophic hypogonadism presented in males during adolescent as delay puberty or as arrested puberty at Tanner stage 3. Rare manifestation of X-linked AHC can be delay onset adrenal insufficiency, partial HH or infertility. Heterozygous females have presentation of Adrenal insufficiency or Hypogonadotrophic hypogonadism [2,5].
Deletion of Xp21 comprising of NR0B1 (X-linked AHC), GK (glycerol kinase deficiency), and in some cases deletion of DMD (Duchenne Muscular Dystrophy). In some cases of Xp21 deletion developmental delay has been reported, when it extends proximally to include deletion of DMD or distally to include IL1RAPL1 and DMD [2,4].
Most often diagnosis of AHC is overlooked as its clinical manifestation is identical to 21-Hydroxylase deficiency. The most note worthy feature of AHC is impaired secretion of androgen, unlike which is observe in CAH [5].
As both conditions are indistinguishable (CAH & AHC), it is of utmost importance that a precise genetic diagnosis is made. Diagnosis of NR0B1-related adrenal hypoplasia congenita is made by detection of either ahemizygous pathogenic variant in NR0B1 or a non-recurrent Xp21 deletion that includes NR0B1 [2,5].
Conclusion
We reported two cases of NR0B1 mutations with dichotomous pubertal presentation. As it is difficult to distinguish AHC from CAH on clinical manifestations because signs and symptoms of both disorders are similar, so it is of utmost importance to distinguish these two condition on precise genetic diagnosis for proper genetic counseling and appropriate and timely management.
References
- Saleem F, Baradhi K. Adrenal Hypoplasia [Internet]. Ncbi.nlm. nih.gov. 2021. [cited 14 December 2021]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549779/
- Achermann J, Vilain E. 2021. NR0B1-Related Adrenal Hypoplasia Congenita. [online] Ncbi.nlm.nih.gov. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1431/ [Accessed 14 December 2021].
- Hormones.gr [Internet]. Hormones.gr. 2021. [cited 14 December 2021]. Available from: http://www.hormones.gr/8488/article/article.html
- Achermann J, Vilain E. NR0B1-Related Adrenal Hypoplasia Congenita [Internet]. Ncbi.nlm.nih.gov. 2021 [cited 14 December 2021]. Available from: https://www.ncbi.nlm.nih.gov/books/ NBK1431/
- Loureiro M, Reis F, Robalo B, Pereira C, Sampaio L. Adrenal hypoplasia congenita: A rare cause of primary adrenal insufficiency and hypogonadotropic hypogonadism. Pediatric Reports. 2015; 7.