
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Left ventricular non-compaction associated with a MYH7 splicing variant (c.818+1G>A)
Juan Gómez1,3,5; Rebeca Lorca1,5; Alejandro Junco Vicente1; Elías Cuesta Llavona3,5; Julián R Reguero1,5; María Martín1; Juan Calvo2; Helena Cigarrán2; Cecilia Corros2; Eliecer Coto1,3,4,5*
1 Unidad de Referencia de Cardiopatías Familiares-HUCA, Genética Molecular y Cardiología, Hospital Universitario Central Asturias, Oviedo, Spain.
2 Servicio Radiología, Hospital Universitario Central Asturias, Oviedo, Spain.
3 Medical Genetics Laboratory, Department of Molecular Medicine, Sapienza University, San Camillo-Forlanini Hospital, Rome, Italy.
4 Departamento de Medicina, Universidad de Oviedo, Oviedo, Spain.
5 Instituto Investigación Sanitaria Principado Asturias-ISPA, Oviedo, Spain.
*Corresponding Author: Eliecer Coto
Hospital Univ. Central Asturias, 33011, Oviedo, Spain.
Email: eliecer.coto@sespa.es
Received : Nov 29, 2021
Accepted : Jan 13, 2022
Published : Jan 20, 2022
Archived : www.jcimcr.org
Copyright : © Coto E (2022).
Abstract
Left Ventricular Noncompaction (LVNC) is a cardiac disease characterized by a trabecular meshwork and deep intertrabecular myocardial recesses that communicate with the left ventricular cavity. MYH7 splicing mutations are very rare, and seem to be restricted to patients with myopathy of LVNC. We characterized at the Cardiac Magnetic Resonance (CRM) and genetic levels in one individual (index case) from a family with LVNC. The patient was heterozygous carrier of a splicing MYH7 intron 8 variant (c.818+1G>A). One of his sons was also mutation carrier but clinically asymptomatic. However, the CRM showed the presence of LVNC. We concluded that the rare MYH7 intron 8 splicing mutation was associated with typical LVNC lesions in the CRM, albeit mutation carriers remain clinically asymptomatic.
Keywords: left ventricular non-compaction; MYH7 gene; RNA decay; cardiac magnetic resonance.
Citation: Gómez J, Lorca R, Vicente AJ, Llavona EC, Reguero JR, et al. Left ventricular non-compaction associated with a MYH7 splicing variant (c.818+1G>A). J Clin Images Med Case Rep. 2022; 3(1): 1597.
Introduction
Left Ventricular Noncompaction (LVNC) is characterised by numerous prominent trabeculations and deep intertrabecular recesses that communicate with the left ventricular cavity but not with the coronary circulation [2]. The clinical manifestation of LVNC ranges from no symptoms to severe symptoms with cardiac arrhythmia, congestive heart failure, and Sudden Cardiac Death (SCD) [25]. The prevalence of LVNC has been reported in the range 0.014-1.3%, although the use of Cardiac Magnetic Resonance (CMR) has increased the detection of morphological features of LVNC [20,21].
LVNC may be familial (inherited) or sporadic (non-familial), the latter diagnosed when LVNC is proven absent in relatives. This sporadic form of LVNC can be acquired, as in highly-trained athletes [7]. A combined molecular testing and cardiological family screening has estimated that around 65% of LVNC cases have a recognised genetic cause [11].
The screening of candidate genes in LVNC patients identified putative pathogenic variants in several genes, including LMNA, ZASP, and DTNA [Ichida et al. 2001]. In addition, some patients might harbour mutations in the cardiac sarcomeric MYH7, MYBPC3, ACTC, TNNT2, TPM1 genes [15,3,22,26,19]. The screening of these genes would facilitate the diagnosis of LVNC cases and the familial genetic counselling [23]. These LVNC genes are frequently mutated in cases with hypertrophic or dilated cardiomyopathies (HCM, DCM). LVNC shares morphologic features with HCM. While HCM can mimics LVNC by the presence of trabeculation and myocardial crypts, LVNC can also present with increased wall thickness [1,13,24,6,12,10]. Moreover, both diseases can occur in the same patient, and in some families the same causative mutation can manifest with either HCM and LVNC phenotypes [14]. These findings supported a common genetic origin for different cardiomyopathic phenotypes.
Most of the reported MYH7 mutations were missense amino acid changes. MYH7 non-sense, frame shifting or splicing mutations are rarely found in patients with cardiomyopathies, suggesting a deleterious effect for this type of severe changes and a strong negative survival effect among mutation carriers. In contrast, these type of mutations are common in the MYBPC3 and other sarcomere related genes [4,8]. One exception was c.818+1G>A, a splicing change in the first nucleotide of intron 8. This change was the first reported MYH7 splice-site mutation, and was found in non-related LVNC patients [15,17].
We present the clinical study of a LVNC patient harbouring this mutation, and provide evidence for the presence of LVNC imaging in the absence of clinical symptoms.
Case presentation
The index case was a 45 years old male that attended our Cardiology Department by suffering ventricular extrasystoles at the age of 33 years. The echography showed a non-compacted dilated cardiomyopathy, and the Cardiac Magnetic Resonance (CMR) a dilated left-ventricule with severe depressed ventricular function (28%), a marked myocardial trabeculation at the medio-ventricular, apical, and apex levels of the left-ventricle wall, with an end diastolic non compacted / compated ratio was > 2.3 (Figure 1). This image is diagnosis of LVNC [13].
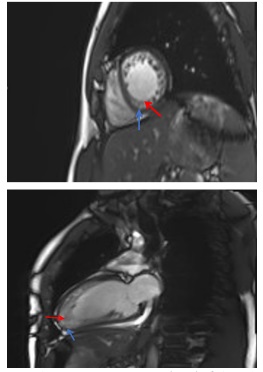
The maternal grandmother and the mother were also affected by non-compacted cardiomyopathy, although they died from non-cardiac cause. We can thus assume the familial dominant inheritance of the disease. The index case has two children (12 and 18 years old), both asymptomatic. The patient was informed about the likely genetic origin of his disease, and signed an informed consent for the genetic study.
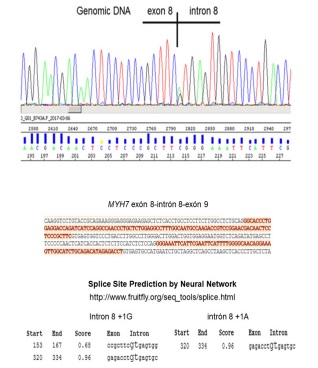
The main LVNC genes (Table 1) were Next Generation Sequenced with semiconductor chips and the Ion Torrent PGM, as reported [8,9]. The only candidate variant found in the 33 sequenced genes was c.818+1G>A, at the splice donor site of intron 8 of MYH7. Other variants were rare or common polymorphisms, and classified as non-pathogenic. To confirm the MYH7 c.818+1G>A variant we Sanger sequenced an exon 7-9 polymerase chain reaction (PCR fragment (Figure 2). This nucleotide change was predicted to eliminate the intron 8 donor site with a bioinformatic tool (Splice Site Prediction by Neural Network; http://www.fruitfly.org/seq_tools/splice.html) (Figure 2). This variant was not found in our screening of 448 HCM patients and 450 healthy controls [Gómez et al, 2017]. Based on the bioinformatic analysis, and its absence in our population as well as in the exome databases, we classified this MYH7 variant as likely pathogenic.
Table 1:Genes (n=33) sequenced in the LVNC patient.
ACTC1 |
LDB3 |
PRDM16 |
ACTN2 |
LMNA |
RBM20 |
ANKRD1 |
MIB1 |
RYR2 |
BAG3 |
MYBPC3 |
TAZ |
DMD |
MYH6 |
TCAP |
DNAJC19 |
MYH7 |
TMPO |
DTNA |
MYL2 |
TNNC1 |
FHL1 |
MYL3 |
TNNI3 |
FLNC |
MYPN |
TNNT2 |
HCN4 |
NKX2-5 |
TPM1 |
LAMA4 |
PLN |
VCL |
We determined this putative mutation in the two patient´s children, and one of them was also c.818+1G>A carrier. He was asymptomatic at the age of 18 years, and the ECG was not conclusive of heart structural abnormalities. This individual was then studied through CMR that showed a slightly dilated leftventricle with myocardial trabeculation at the anterolateral, inferolateral, anteroseptal, apical, and apex levels and non compaction criteria. No signs of cardiac hypertrophy or fibrosis were observed. The right-ventricle and the two atrium were normal in size and structure. The left ventricle systolic function was also normal.
Discussion
Imaging features of HCC on MRI
Most of the reported MYH7 mutations found in cardiomyopathies were missense amino acid changes. In our screening of 448 HCM patients we identified a total of 48 MYH7 likely pathogenic variants, and none of them was predicted to affect RNA splicing [9]. At least one MYH7 splicing mutation (intron 38 +1 G>A) has been reported in a patient with early onset myopathy (8 years old) and symptoms of LVNC [5]. In contrast, our patient harbouring the intron 8 +1 G>A change had no symptoms of myopathy, and myopathy was also absent in the cases harbouring the same mutation [15]. It thus seems that the site of the splicing mutation relative to the MYH7 transcript could define the presence of skeletal symptoms or a pure cardiac phenotype. This observation is not unprecedented, because mutations in the MYH7 gene can result in either Laing-type early-onset distal myopathy or HCM, suggesting that mutations affecting different protein domains can result in different clinical outcomes [18].
Klassen et al. found the MYH7 intron 8 splicing mutation in a total of nine patients from 2 families, with all them fulfilling the morphological criteria for LVNC even at young age (8 years). They concluded that this mutation was associated with a high morphological penetrance, although some of the carriers remained clinically asymptomatic. The mother of our patient was also affected, and she could be also a carrier of the intron 8 variant, although she was not available for the genetic study. Moreover, in agreement with Klassen et al. one of our index case sons was also mutation carrier and remained clinically asymptomatic at the age of 18. However, the CMR was conclusive of LVNC, that supports a high penetrance for this mutation at the morphological level.
Conclusion
We present a family with left-ventricular non-compation and carrier of a splicing nucleotide change in intron 8 of the MYH7 gene. This mutation was associated with a LVNC image in the absence of clinical symptoms.
Declarations
Funding sources: This work was supported by grant of the Spanish Ministerio de Economía y Competitividad-Instituto de Salud Carlos III and the Fondos Europeos de Desarrollo Regional (FEDER funds) (grant PI18/00719).
Competing interests: All the authors declare no conflict of interest relative to this work.
References
- Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: A distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol. 2014; 64: 1840-1850.
- Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R, et al. Isolated noncompaction of left ventricular myocardium: A study of eight cases. Circulation. 1990; 82: 507–513.
- Dellefave LM, Pytel P, Mewborn S, Mora B, Guris DL, Fedson S, et al. Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ Cardiovasc Genet. 2009; 2: 442-449.
- Erdmann J, Daehmlow S, Wischke S, Senyuva M, Werner U, Raible J, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet. 2003; 64: 339–349.
- Fiorillo C, Astrea G, Savarese M, et al. MYH7-related myopathies: Clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis. 2016; 11: e91.
- Frischknecht BS, Attenhofer Jost CH, Oechslin EN, et al. Validation of noncompaction criteria in dilated cardiomyopathy, and valvular and hypertensive heart disease. J Am Soc Echocardiogr. 2005; 18: 865–872.
- Gati S, Chandra N, Bennett RL, Reed M, Kervio G, Panoulas VF, et al. Increased left ventricular trabeculation in highly trained athletes: Do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart. 2013; 99: 401-408.
- Gómez J, Reguero JR, Morís C, et al. Mutation analysis of the main hypertrophic cardiomyopathy genes using multiplex amplification and semiconductor next-generation sequencing. Circ J. 2014; 78: 2963-2971.
- Gómez J, Lorca R, Reguero JR, et al. Screening of the Filamin C Gene in a Large Cohort of Hypertrophic Cardiomyopathy Patients. Circ Cardiovasc Genet. 2017; 10: pii: e001584.
- Haland TF, Saberniak J, Leren IS, Edvardsen T, Haugaa KH. Echocardiographic comparison between left ventricular noncompaction and hypertrophic cardiomyopathy. Int J Cardiol. 2017; 228: 900-905.
- Hoedemaekers YM, Caliskan K, Michels M, Frohn Mulder I, van der Smagt JJ, Phefferkorn JE, et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet. 2010; 3: 232-239.
- Ichida F, Tsubata S, Bowles KR, et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001; 103: 1256–1263.
- Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart. 2001; 86: 666–671.
- Kelley Hedgepeth A, Towbin JA, Maron MS. Maron, Images in cardiovascular medicine. Overlapping phenotypes: Left ventricular noncompaction and hypertrophic cardiomyopathy. Circulation. 2009; 119: e588–e589.
- Klaassen S, Probst S, Oechslin E, et al. Mutations in sarcomere protein genes in left ventricular non-compaction. Circulation. 2008; 117: 2893–2901.
- Maron MS, Rowin EJ, Lin D, et al. Prevalence and clinical profile of myocardial crypts in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2012; 5: 441–447.
- Mazzarotto F, Hawley MH, Beltrami M, et al. Systematic largescale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet Med. 2021; 23: 856-864.
- Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am. J. Hum. Genet. 2004; 75: 703–708.
- Nijak A, et al. Left ventricular non-compaction with Ebstein anomaly attributed to a TPM1 mutation. Eur J Med Genet. 2017; S1769-7212: 30449-30454.
- Oechslin E, Jenni J. Left ventricular non-compaction revisited: A distinct phenotype with genetic heterogeneity? Eur Heart J. 2011; 1446–1456.
- Petersen SE, Selvanayagam JB, Wiesmann F, Robson MD, Francis JM, Anderson RH, et al. Left ventricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. 2005; 46: 101–105.
- Probst S, Oechslin E, Schuler P, Greutmann M, Boye P, Knirsch W, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011; 4: 367-374.
- Shieh JT. Implications of genetic testing in noncompaction/ hypertrabeculation. Am J Med Genet C Semin Med Genet. 2013; 163C: 206-211.
- Stöllberger C, Finsterer J. Left ventricular hypertrabeculation/ noncompaction. J Am Soc Echocardiogr. 2004; 17: 91–100.
- Towbin JA. Left ventricular noncompaction: A new form of heart failure. Heart Fail. Clin. 2010; 453–469.
- Wessels MW, Herkert JC, Frohn Mulder IM, et al. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet. 2015; 23: 922-928.