
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
A novel nonsense MPS VI mutation in a south Moroccan patient
Es-said Sabir1 ; Naima Marzouki2 ; Karima Lafhal1 ; Rabiy Elqadiry3 ; Najwa Imad4 ; Nadia El idrissi Slitine4 ; Mounir Bourrous5 ; Noureddine Rada3 ; Mohamed Bouskraoui3 ; Naima Fdil1
1Metabolics Platform, Biochimistry Laboratory, Faculty of Medicine, Cadi Ayyad University, Marrakech, Morocco.
2 Medical Biology Laboratory, Mohammed VI University Hospital, Marrakech, Morocco.
3 Department of Pediatrics, Mohammed VI University Hospital, Faculty of Medicine and Pharmacy, University, Cadi Ayyad, Marrakech, Morocco.
4 Neonatal Intensive Care Department, Mohammed VI University Hospital, Marrakech, Morocco.
5 Pediatric Emergency Department, Mohammed VI University Hospital, Marrakech, Morocco.
*Corresponding Author: Naima Fdil
Metabolics Platform, Biochemistry Laboratory, Faculty of Medicine, Cadi Ayyad University, SidiAbbad,
BP 40000, Marrakech, Morocco.
Email: nfdil@yahoo.fr
Received : Dec 06, 2021
Accepted : Jan 19, 2022
Published : Jan 26, 2022
Archived : www.jcimcr.org
Copyright : © Fdil M (2022)
Abstract
A 12-years-old, Moroccan child with a history of skeletal deformities, gait disorder, Short stature, valvulopathy, facial dysmorphy, corneal opacity, umbilical hernia, and hepatomegaly. He has onset from birth, delayed first milestones (setting, gait, and language) and severe dysostosis multiplex. Elevated urinary glycosaminoglycans concentration was found, enzymatic assays show a low level of ARSB, genetic testing confirmed a diagnosis of Mucopolysaccharidosis type VI with a novel mutation in ARSB gene. There is a homozygous frameshift nonsense mutation by two thymine deletion in 770 and 771 codons (c. [770_771del TT]; [770_771del TT]). This is the first case and mutation of Mucopolysaccharidosis type VI detected in south Morocco.
Keywords: : mucopolysaccharidosis VI; ARSB gene; nonsense mutation; ERT
Citation:Sabir ES, Marzouki M, Lafhal K, Elqadiry R, Fdil M, et al. A novel nonsense MPS VI mutation in a south Moroccan patient. J Clin Images Med Case Rep. 2022; 3(1): 1614.
Introduction
Mucopolysaccharidosis VI (MPS VI) or Maroteaux–Lamy syndrome (OMIM 253200) is a rare genetic disease first described in 1963 by the French doctors Pierre Maroteaux and Maurice Lamy [1]. The disease inherited as an autosomal recessive trait and caused by mutations in the ARSB gene that encodes the lysosomal enzyme ARSB [2]. This gene, located in chromosome 5 (5q13-5q14), presents 408 variants (Including filtered: 445), five copy number Variant (CNVs including filtered: 47) and UCSC Browser 5: 78073032-78281910. More than 148 ARSB mutations were reported, causing absent or reduced arylsulfatase B (N-acetylgalactosamine 4-sulfatase) activity and interrupted dermatan sulfate and chondroitin sulfate degradation, this enzyme is involved in the catabolism of Glycosaminoglycan (GAG) Dermatan Sulfate (DS) [3]. Deficient activity of Arylsulfatase B (ARSB) leads to progressive accumulation of DS and affects connective tissues of the skin, heart valves, airways, and the skeleton [4,5].
Progressive Accumulation of Glycosaminoglycans (GAGs) in organs and tissues leads to the development of multisystem clinical manifestations. The presentation of MPS VI is genotypically and phenotypically diverse, with a large number of potential disease-causing mutations and a phenotypic spectrum ranging from very slow to very rapid progressing disease. Diagnosis generally requires evidence of clinical phenotype, arylsulfatase B enzyme activity <10% of the lower limit of normal and demonstration of a normal activity of a different sulfatase enzyme (to exclude multiple sulfatase deficiency). The finding of elevated urinary dermatan sulfate with the absence of heparan sulfate is supportive [6,7]. Evidence of MPS VI diagnosis supported in our patient by clinical exam and analyzed tests, as we will describe in this paper. Informed consent was read and signed by the patient’s family.
Materials & methods
Sample collection and preparation An 11-years-old male boy with the clinical signs of a possible inborn error of metabolism was referred to our laboratory. Morning urine sample, Dried Blood Spot (DBS), and Ethylene Diamine Tetra Acetic Acid (EDTA) blood were collected.
Urinary GAG analysis
The quantitative determination of urinary GAGs based on the spectrometric determination of the binding of Glycosaminoglycan’s (GAGs) with 1,9-dimethylen blue, in morning urine sample [8,9] was performed using ultraviolet-visible spectrophotometry on a UV-visible 1601 Shimadzu spectrophotometer. The final GAG results are expressed in mg/mmol creatinine and the reference values depend on age.
Lysosomal enzyme assays
The activities of four lysosomal enzymes, α-L-iduronidase (IDUA) (EC 3.2.1.76 for MPS I), iduronate-2-sulphate sulphatase (EC 3.1.6.13 for MPSII), β-D-galactosidase (EC 3.2.1.23 for MPS IVB), and Arylsulphatase B (EC3.1.6.12 for MPS VI), were assayed by previously established fluorescence methods [10,11].ARSB gene sequencing
DNA extraction, PCR and Sequencing of all exons and introns flanking the ARSB gene were done. The sequencing results were analyzed and compared to the published human genome sequence (NCBI Genbank, Gene ID: 3425, RefSeq # NM_000203.3 (ENST00000247933), http://www.ncbi.nlm.gov) and Ensemble: (http://www.ensembl.org).
Clinical history
This is the case of a child (born on 05.03.2006), who consulted for the first time on 29.05.2008 in the pediatrics department, he is the fourth of five siblings, originally from Marrakech. He comes from a consanguineous marriage of 3rd degree with no similar cases in the family. The pregnancy estimated at term. Delivered at home, with no medical assistance, and reported without incident. Not breastfed, presenting a psychomotor delay: Sitting position, holding the head and standing at the age of 14 months, walking at the age of 17 months and the language acquisition began after he was 2 years old. His parents report a possible deafness at birth (a baby was indifferent front of a hard noise), a bulge at the age of 1 year, and adenoidectomy at 4 years of age.
The beginning of his symptoms seems to go back to the age of 14 months, by a gait disorder (knee Varus) with deformity of the spine, which motivated a transfer to pediatric surgery.
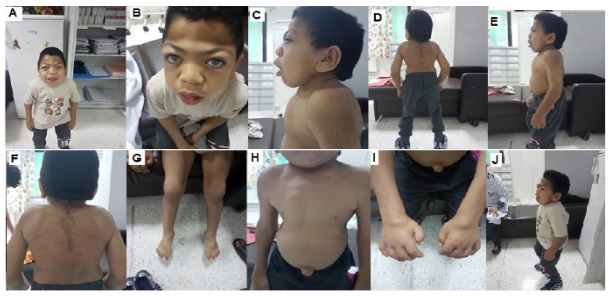
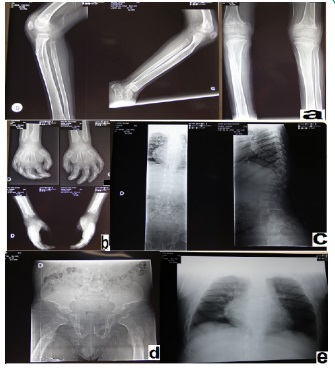
On examination, the patient was conscious, T° 37°C, weight 11 kg and height of 82 cm. He presented a facialdysmorphy (flattened nose, thick cheeks and lips, hypertelorism, macroglossia, detached earlobes and diffuse Rhonchi lung sounds). Osteon - articular exam showed early scoliosis, lumbar kyphosis, scaphocephaly, sternal protrusion, bilateral thoracic depression, knee varum and flat feet (Figure 2). Hehada hepatomegaly and an umbilical hernia of 1.5 cm, other hernia orifices were free.
Neurological report showed walking by support, plantar reflexes in flexion, muscle tone and strength preserved. Dermatological findings objectified multiple mongoloid rashes in the back and buttocks and next to the left knee. Ophthalmological analysis found corneal opacity.
A discomfort at the age of 2 years (01.03.2008) required a cardiac exam that demonstrated a BAV at ECG and aortic valve involvement (thickened valve) by echography. He develops cardiac valve insufficiency and stenosis. Cerebral CT Scan (15.05.2014) demonstrated bilateral and symmetrical hypo density of the white matter that related to his metabolic disease.
The patient was 10 years old on 09.12.2016, Weight: 21 kg (-2.5 DS), Size: 102 cm (-5 DS). Clinical exam detect bilateral cryptorchidism and audiogram revealed bilateral deafness.
Results
Our patient on 17.05.2018 was 12 years old. He was smiling, cooperating and kind. Nevertheless, there was a worsening of the symptoms described before (facial dysmorphy, skeletal deformities, apnea and corneal opacity). There were also repetitive infections (atopy) that made the patient and his family uncomfortable.
GAGs concentrations (96, 83 mg/mmol creatinine) was higher than normal valor: 6, 7-15, 5 mg/mmol. The activity of arysulfatase B is below its reference range (Table 1). This may be in agreement with an MPS VI. The other enzyme activities Alpha-iduronidase (MSMS), Idoronate-2-sulfatase, Beta-galactosidase are within their respective reference ranges or deviate just slightly from those. Thus, there is no indication of MPSI and MPSII.
Table 1:Enzyme activities in dried blood spot sample of the patient.
Lysosomal enzymes from dried blood |
Result |
Reference range |
Alpha-iduronidase |
466.94 |
200-2614 pmol/spot*20 h |
The following homozygous detected mutation: c. [770_771delTT]; [770_771delTT] confirms MPSVI disease in our patient. This is homozygous deletion of two nucleotides leads to a frameshift (nonsense mutation). Variant described using the Human Gene Variant Sequence (HGVS) nomenclature. Common benign variants in the gene identified. Nevertheless, not have been included in this report as do not influenced phenotype disease.
Table 1:Enzyme activities in dried blood spot sample of the patient.
Discussion
In homogeneous populations such as Morocco’s, which has a high rate of consanguineous marriages, there is an increased rate of autosomal recessive genetic disorders such as MPS VI. It is a rare progressive lysosomal storage disorder caused by the deficit of the ARSB enzyme. According to international studies, no incidence data for this MPS VI mutation published before.
We described here a new homozygous nonsense mutation of the ARSB gene in a south Moroccanchild. This result confirms MPS VI diagnosis. This mutation located in domain 1 (S37-R388) at exon 4 of ARSB gene. The two-thymine deletion engender a non-sense mutation leading to premature stop codon and lack of the entire region after first 770 nucleotide. In genetics, a point-nonsense mutation is a point mutation in a sequence of DNA that results in a premature stop codon, or a point-nonsense codon in the transcribed mRNA, and in a truncated, incomplete, and usually nonfunctional protein product. The effect of a point-nonsense mutation depends on the proximity of the point-nonsense mutation to the original stop codon, and the degree to which functional subdomains of the protein are affected. Despite an expected tendency for premature termination codons to yield shortened polypeptide products, in fact the formation of truncated proteins does not occur often in vivo. Many organisms—including humans and lower species, such as yeast—employ a point-nonsense-mediated mRNA decay pathway, which degrades mRNAs containing point-nonsense mutations before they can translated into nonfunctional polypeptides. The truncated mRNA lacks more than half of the original mRNA (part of exon 4 and the exons [5-8]. Moreover, it results in loss of enzyme activity. Which explain severe and rapid disease evolution in our patient.
In other hand, delay diagnosis had a negative impact on clinical evolution of our patient. He did not benefit from early enzyme treatment. The development of a routine genetic analysis for this type of hereditary metabolic diseases is necessary to improve the vital prognosis and outcome of these patients. The prevalence pattern of mucopolysaccharidosis VI in Morocco is not determined, based on data from our Laboratory, among 38 diagnosed patients with different types of MPS [6,7]; 3 patients were diagnosed with severe MPSVI phenotype.
Conclusion
Physician awareness for early diagnosis and treatment is the only recourse to avoid a serious complication of MPS VI and to improve life quality of patients and their families.
Acknowledgment: The authors appreciate family members who kindly contributed in this study.
References
- Black SH, Pelias MZ, Miller JB, Blitzer MG, Shapira E. MaroteauxLamy syndrome in a large consanguineous kindred: Biochemical and immunological studies. Am J Med Genet. 1986; 25: 273- 279.
- Maroteaux P, Leveque B, Marie J, Lamy M. A New Dysostosis with Urinary Elimination of Chondroitin Sulfate B. La Pressemedicale. 1963; 71: 1849-1852.
- De Almeida-Barros RQ, de Medeiros PFV, de Almeida Azevedo MQ, de Oliveira Lira Ortega A, Yamamoto ATA, Dornelas SKL, Bento PM. Evaluation of oral manifestations of patients with mucopolysaccharidosis IV and VI: Clinical and imaging study. Clin Oral Investig. 2018; 22: 201–208.
- Lin WD, Lin SP, Wang CH, Hwu WL, Chuang CK, et al. Genetic analysis of mucopolysaccharidosis type VI in Taiwanese patients. ClinChim Acta. 2008; 394: 89–93.
- Yassaee VR, Hashemi Gorji F, Miryounesi M, Rezayi A, Ravesh Z, et al. Clinical, biochemical and molecular features of Iranian families with mucopolysaccharidosis: A case series. Clin Chim Acta. 2017; 474: 88–95.
- Fdil N, Sabir ES, Ezoubeiri A, Elqadiry R, Daoudi A, Lalaoui A, et al. Implementation of an affordable method for MPS diagnosis from urine screening to enzymatic confirmation: Results of a pilot study in Morocco. 2020.
- Sabir ES, Lafhal K, Ezoubeiri A, Harkati I, Sbyea S, Aldámiz‐Echevarría L, Fdil N. Usefulness of urinary glycosaminoglycans assay for a mucopolysaccharidosis‐specific screening. Pediatrics International. 2020; 62: 1077-1085.
- Laura López-Marín, Luis G Gutiérrez-Solana, Luis Aldamiz-Echevarria Azuara, Rogelio Simón de las Heras Anna Duat Rodríguez, Verónica Cantarín Extremera. Detection by Urinary GAG Testing of Mucopolysaccharidosis Type II in an At-Risk Spanish Population. JIMD Rep. 2013; 10: 61–68.
- Fernando Andrade José, Angel Prieto, Javier Elorz, Sergio Martín, Pablo Sanjurjo, Luis Aldámiz-Echevarría. Stability of urinary glycosaminoglycans in patients with mucopolysaccharidoses. Clinica Chimica Acta. 2008; 388: 73-77.
- Chamoles NA, Blanco MB, Gaggioli D, Casentini C. Hurler-Like phenotype: Enzymatic diagnosis in dried blood spots on filter paper. Clin Chem. 2001; 47: 2098–2102.
- Voznyi YV, Keulemans JLM, Beyer EM, Van Diggelen OP. A fluorogenic assay for the diagnosis of Hunter disease (MPS II). J Inherit Metab Dis. 2001; 24: 675–680.