
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Research Article - Open Access, Volume 3
Genotype-phenotype correlations for the type 1 lissencephaly of eight Tunisian children
Meriam Hadj Amor1,2; Nejla Soyah3; Nathalie Drouot4; Ichraf Kraoua5; Hela Ben Khelifa1; Wafa Slimani1,2; Sarra Dimassi1,6; Hanene Hannachi1; Ahlem Attig1; Hedia Klai5; Adnene Mlika3; Lamia Boughamoura3; Khaled Ben Helel7; Ilhem Turki5; Naziha Gouider Khouja5; Jamel Chelly4,8; Ali Saad1,6; Soumaya Mougou Zerelli1,6*
1 Department of Human Cytogenetics, Molecular Genetics and Reproductive Biology, Farhat Hached University Teaching Hospital, Sousse, Tunisia.
2 High Institute of Biotechnology, Monastir University, Monastir, Tunisia.
3 Pediatric department, Farhat Hached University Teaching Hospital, Sousse, Tunisia.
4 Institute of Genetics and Molecular and Cellular Biology, Illkirch, France.
5 Department of Child and Adolescent Neurology, National Institute Mongi Ben Hmida of Neurology, Tunis, Tunisia.
6 Common Service Units for Research in Genetics, Faculty of Medicine of Sousse, University of Sousse, Sousse, Tunisia.
7 Pediatric department, Ibn Jazzar University Teaching Hospital, Kairouan, Tunisia
8 Laboratory of Medical Genetics, Strasbourg University Hospital, Strasbourg, France.
*Corresponding Author: Soumaya Mougou Zerelli
Human Cytogenetics, Molecular Genetics and Reproductive Biology, Farhat Hached University Teaching Hospital,
Ibn El Jazzar street, Sousse 4000, Tunisia.
Email: mougousoumaya@yahoo.fr
Received : Nov 29, 2021
Accepted : Feb 02, 2022
Published : Feb 09, 2022
Archived : www.jcimcr.org
Copyright : © Zerelli SM (2022).
Abstract
Background: Lissencephaly represents a rare subgroup of genetically distinct neurological disorders of neuronal migration characterized by a paucity or absence of cerebral gyration. The most common form of lissencephaly has been isolated and referred to as classic or type 1 lissencephaly. It is frequently related to abnormalities within LIS1 or DCX genes, with abnormalities ranging from single base pair substitutions to contiguous gene deletions.
Methods: In this study, we report, for the first time, a clinical and genetic characterization of eight unrelated Tunisian children presenting type 1 lissencephaly. We screened LIS1 and DCX abnormalities thanks to a combination of molecular cytogenetic methods and next generation sequencing.
Results: One deletion of DCX and three deletions of LIS1 were observed. One of these deletions was inherited from a maternal reciprocal translocation and estimated to approximately 2,9 Mb length. In addition, a 26 Kb LIS1 deletion was detected and refined between exon 3 up to exon 11 by target capture and sequencing. The last LIS1 rearrangement was a mutation (c.779T>A, p.V260E). Finally, two novel DCX mutations were found out (c.910G>C, p.G304R/c.436T>C, p.F146L).
Conclusions: Our data confirm the individuality and originality of type 1 lissencephaly on both the phenotypic and the genetic levels. Furthermore, our data confirm again that LIS1 and DCX are the most genes associated with type 1 lissencephaly and spotlight the usefulness of developing approaches and methods for detecting a large number of known causative gene mutations.
Keywords: DCX; LIS; molecular cytogenetic methods; type 1 lissencephaly; next generation sequencing.
Abbreviations: LIS1: Lissencephaly 1; DCX: Doublecortin; SMD: Syndrome de Miler Dieker; ILS: Isolated Lissencephaly.
Citation: Amor MH, Soyah N, Drouot N, Kraoua I, Zerelli SM, et al. Genotype-phenotype correlations for the type 1 lissencephaly of eight Tunisian children. J Clin Images Med Case Rep. 2022; 3(2): 1645.
Introduction
Neuronal migration is an extremely complex process, in which neurons move away from their original site in the ventricular zones to their destination in specific brain regions [1]. The most common neuronal migration disorder is type 1 lissencephaly, caused by defects in neurogenesis and nucleokinesis [2]. It is characterized by paucity (agyria) or the absence of cerebral gyration (pachygyria) resulting in a “smooth brain” [3]. Lissencephalic patients have various clinical manifestations, including severe psychomotor retardation and epilepsy. In addition, neurological deficits and an early death were observed [4].
The heterogeneity of the associated genes to lissencephaly has historically made it difficult to determine the specific etiology. To the best of our knowledge, 25% are characterized at the molecular level [5]. Nevertheless, LIS1 and DCX are the major candidate genes. PAFAH1B1, a platelet-activating factor acetylhydrolase isoform 1B α subunit, also known as LIS1, is involved in a signal transduction pathway that is important for cerebral development [6-7]. In fact, LIS1 deletions extending from singleexon deletions to deletions of the entire coding region cause a more severe phenotype than missense mutation. Besides, LIS1 mutations are responsible for about 65% of type 1 lissencephaly [8], while mutations in DCX, a gene involved in microtubules stabilization, are typically associated with classic lissencephaly in males with a prevalence of 25% [9].
In this study, we report the clinical and molecular genetic findings in eight Tunisian children having type 1 lissencephaly. One DCX deletion and four LIS1 deletions were found and were predicted to result in truncated protein. In addition, a novel mutation in LIS1 and DCX genes were detected and were predicted to result in non-functional protein.
Interestingly, our data contributes to further defining the molecular characteristics associated with type 1 lissencephaly to improve the genotype-phenotype correlation.
Patients and methods
Patients
Eight children were referred to our department of Cytogenetics and Biology of Reproduction for their developmental delay and dysmorphic features. The clinical details of each patient are shown in Table 1. Photographs of patients 2, 3, 5 and 7 are shown in Figure 1. There was no history of lissencephaly in any of the eight patients except for patient 3. The Magnetic Resonance Imaging (MRI) of patients 1 and 2 are shown in Figure 2.
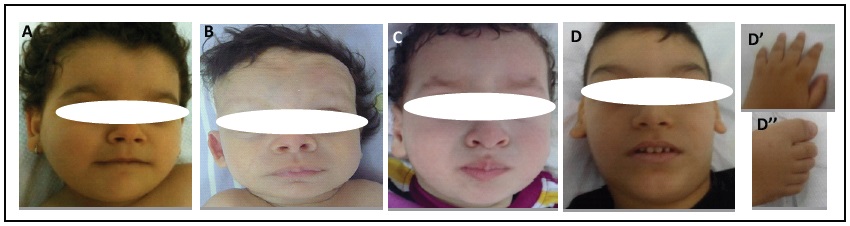
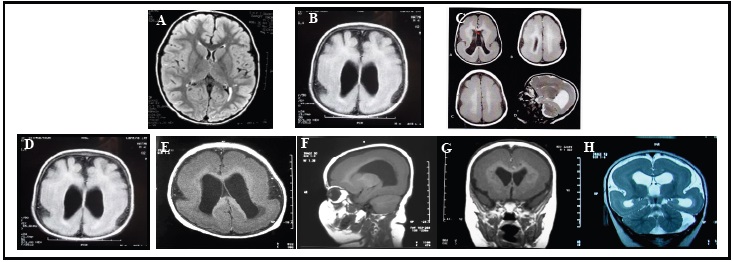
Table 1: Clinical and molecular characteristics of eight patients with lissencephaly spectrum.
Patient |
Sex |
Age |
Clinical examination |
Neurological examination |
Brain MRI |
Genetic data |
Inheritance |
In silico analyses |
||
FISH analysis |
CGH analysis |
Target capture |
||||||||
P1 |
M |
14 m |
Psychomotor retardation, severe and early epilepsy, moderate facial dysmorphia |
Global hypotonia |
Posterior agyria, anterior pachygyria with ventriculomegaly |
46,XY,ish del(17p13.3)(LIS1*1) |
Not done |
Not done |
Father |
Not done |
P2 |
F |
4 y |
Psychomotor retardation, West syndrome |
Axial and peripheral hypotonia |
Scattered agyria with ventriculomegaly |
46,XX,ish del(17p13.3)(LIS1*1) |
Not done |
Not done |
De novo |
Not done |
P3 |
F |
4 m |
facial dysmorphia, epilepsy, growth retardation |
Axial hypotonia |
Type 1 lissencephaly withleukomalacia |
46,XX,der(17)t(3;17)(p26.2;p13.3)mat |
46,XX.arr[GRCh18]3p26.2(224727_3864822)X3,17p13.3(48539_2976723)X1 mat |
Not done |
Mother |
Not done |
P4 |
M |
1 y and 8 m |
Psychomotor retardation, epilepsy, West typique syndrome, facial dysmorphia |
Axial and peripheral hypotonia |
Type 1 lissencephaly (Agyria) and corpus callosum agenesis |
LIS1*2 |
46,XY.arr[GRCH18]17p13.3(2522672-2549373)x1 |
Deletion from exon 3 up to exon 11 of LIS1 gene |
Not done |
Not done |
P5 |
M |
6 y and 1/2 |
West syndrome, brachydactyly, facial dysmorphia, growth retardation, psychomotor retardation |
Axial hypotonia |
Diffuse pachygyria |
LIS1*2 |
arr(1-22)(XY)x2 |
LIS1 mutation (c.779T>A, |
De novo |
Mutation Taster: Diseasecausing (0,99) |
P6 |
M |
6 y |
NA |
NA |
Type 1 lissencephaly |
46,XY,ish del(Xq23)(DCX--) |
Not done |
Not done |
NA |
Not done |
P7 |
M |
13 m |
West syndrome, facial dysmorphia, epilepsy |
Axial and peripheral hypotonia |
Agyria, atrophy of the white matter, corpus callosum hypoplasia, ventriculomegaly |
LIS1*2 |
Not done |
DCX mutation (c.910G>C, |
De novo |
Mutation Taster: Diseasecausing (0,99) |
P8 |
M |
15 y |
Facial dysmorphia, mental retardation, speech delay, walking delay |
Normal |
Type 1 lissencephaly |
LIS1*2 |
Not done |
DCX mutation (c.436T>C,
|
Mother |
Mutation Taster: Diseasecausing (0,99) |
tient |
Sex |
Age |
Clinical examination |
Neurological examination |
Brain MRI |
Genetic data |
Inheritance |
In silico analyses |
||
FISH analysis |
CGH analysis |
Target capture |
||||||||
P1 |
M |
14 m |
Psychomotor retardation, severe and early epilepsy, moderate facial dysmorphia |
Global hypotonia |
Posterior agyria, anterior pachygyria with ventriculomegaly |
46,XY,ish del(17p13.3)(LIS1*1) |
Not done |
Not done |
Father |
Not done |
F |
4 y |
Psychomotor retardation, West syndrome |
Axial and peripheral hypotonia |
Scattered agyria with ventriculomegaly |
46,XX,ish del(17p13.3)(LIS1*1) |
Not done |
Not done |
De novo |
Not done |
|
P3 |
F |
4 m |
facial dysmorphia, epilepsy, growth retardation |
Axial hypotonia |
Type 1 lissencephaly withleukomalacia |
46,XX,der(17)t(3;17)(p26.2;p13.3)mat |
46,XX.arr[GRCh18]3p26.2(224727_3864822)X3,17p13.3(48539_2976723)X1 mat |
Not done |
Mother |
Not done |
P4 |
M |
1 y and 8 m |
Psychomotor retardation, epilepsy, West typique syndrome, facial dysmorphia |
Axial and peripheral hypotonia |
Type 1 lissencephaly (Agyria) and corpus callosum agenesis |
LIS1*2 |
46,XY.arr[GRCH18]17p13.3(2522672-2549373)x1 |
Deletion from exon 3 up to exon 11 of LIS1 gene |
Not done |
Not done |
P5 |
M |
6 y and 1/2 |
West syndrome, brachydactyly, facial dysmorphia, growth retardation, psychomotor retardation |
Axial hypotonia |
Diffuse pachygyria |
LIS1*2 |
arr(1-22)(XY)x2 |
LIS1 mutation (c.779T>A, |
De novo |
Mutation Taster: Diseasecausing (0,99) |
P6 |
M |
6 y |
NA |
NA |
Type 1 lissencephaly |
46,XY,ish del(Xq23)(DCX--) |
Not done |
Not done |
NA |
Not done |
P7 |
M |
13 m |
West syndrome, facial dysmorphia, epilepsy |
Axial and peripheral hypotonia |
Agyria, atrophy of the white matter, corpus callosum hypoplasia, ventriculomegaly |
LIS1*2 |
Not done |
DCX mutation (c.910G>C, |
De novo |
Mutation Taster: Diseasecausing (0,99) |
P8 |
M |
15 y |
Facial dysmorphia, mental retardation, speech delay, walking delay |
Normal |
Type 1 lissencephaly |
LIS1*2 |
Not done |
DCX mutation (c.436T>C,
|
Mother |
Mutation Taster: Diseasecausing (0,99) |
This study was approved by the local Ethics Board of the University Hospital Farhat Hached. Written informed consents to genetic testing and picture publication were obtained from the parents.
Karyotype
Chromosomal analysis was performed according to standard procedures for all the patients. Metaphase chromosome preparations were obtained by Phytohemagglutinin (PHA) stimulated lymphocyte culture. Cell cultures were incubated for 72 h. Chromosome analysis was carried out applying R-banding at a 500-band level according to the International System of Human Cytogenetic Nomenclature (ISCN) 2016 [10].
Fluorescence In Situ hybridization (FISH)
FISH was performed on blood lymphocytes blocked on metaphases of all the patients, according to the standard protocol. Two probes screening chromosome 17 short arm and chromosome X short arm were used: Commercial probes; Miller-Dieker/Lissencephaly region probe set: LISI (Red) and RARA (Green) (Vysis) (Abbott Laboratories, IL, USA) and DCX probe. Then, probes were co-denaturized for 7 min at 75°C. After overnight hybridization at 37°C and washing, chromosomes were counterstained with a 4,6 Diamino-2-Phenylindole (DAPI). The hybridized chromosomal spreads were analyzed using a fluorescent microscope equipped with appropriate filters and a Cytovision FISH system image capture software (Axioskop Zeiss® fluorescent microscope). Slides were scored on the basis of the number of probe signals for each metaphase. For each target area, ten hybridized metaphases were analyzed.
Array CGH
Oligonucleotide array CGH was performed using the Agilent Human Genome CGH Microarray Kit 44K®. This microarray consisted of more than 44,000 oligonucleotide probes that spanned both coding and non-coding regions. The coverage of the human genome was made with an average spatial resolution of 75,000 pair bases.
The patient’s DNA as well as a reference DNA was fragmented by heat at 95°C for 20 minutes. Each fragmented DNA product was labeled by random priming using either ULS5 or ULS3. After column-purification, probes were denaturized and preannealed with 5 μg of human Cot-1 DNA, 10 μl of CGH Blocking agent and 55 μl of hybridization buffer. Hybridization was performed at 65°C during 24 h. The microarray was washed, scanned and analyzed with Agilent Feature Extraction® 9.1 software. Results were interpreted using DNA analytics® 4.5 software. Only imbalances involving three or more adjacent probes were held. The identification of probes with a significant gain or loss was based on the log2 ratio plot deviation from 0 with cutoff values of 0.5 to 1, and –0.5 to –1, respectively
Panel capture sequencing, alignment, and variant calling
We designed a targeted sequencing panel of 198 genes related to cortical malformation which are listed in Table 1 in the supplement. Gene panel sequencing of patients was performed using Seq Cap EZ Med Exome (Roche Nimble Gene, USA). We manually processed each series of 24 DNA samples corresponding to index cases for library preparation, and then libraries were pooled for capture and enrichment reaction.
For the sequencing step, paired-end read sequences were performed on Illumina Hi Seq 4000 sequencing platforms (Illumina, Inc., San Diego, California, USA).
As for the bioinformatics analysis of sequencing data, sequence quality was assessed with Fast QC, then the reads were mapped using BWA-MEM against the hg19 genome reference, sorted and indexed in a bam file (Samtools 1.4.1), duplicates were flagged (Sambamba) and coverage was calculated (PicardTools). Variant calling was done with GATK Haplotype Caller. Variants were then annotated with Snp Eff, db NSFP, gnom AD, Clin Var, and an internal database.
Sanger sequencing
Potentially pathogenic mutations identified by targeted sequencing panel were confirmed by Sanger sequencing. The sequencing of PCR amplified fragments was performed on an ABI3100 with the Big Dye Sequencing Kit according to the manufacturer’s specifications (Thermo Fischer Scientific, Waltham, MA).
Primers used for PCR were: F: 5’-AGACATCCAGGAACATGCGA-3’ and R: 5’-TTGTCAGACAGAGATCGCGT-3’ (DCX, exon 2), or F: 5’-AAAGTTCTACTCCAGTGTCAGTGTG- 3’ and R: 5’-TGGAGGAAGAGTCCGTCAAC- 3’ (DCX, exon 3), or F: 5’-TCGACTTGCATCTCTAACTTGG-3’ and R: 5’-TGCATTAACAGCCCTGAAAC- 3’ (LIS1, exon 8).
Results
The conventional cytogenetic analysis revealed a normal karyotype in all patients. For these patients, we conducted a FISH using a probe specific for the LIS1 gene on 17p13.3 and DCX gene on Xq23. This technique showed one microdeletion of DCX gene (P6) and three microdeletions of LIS1gene (P1, P2 and P3) (Figure 3). Parents were also analyzed by FISH using a probe specific for LIS1 gene, in order to study the nature of de novo or inherited microdeletion. One of these deletions (P3) was inherited from a maternal reciprocal translocation (t (3; 17) (p26.2; p13.3). Array CGH analysis estimated the loss of at least 2,9 Mb on the short arm of chromosome 17 (Figure 3). Interestingly, a 26KbLIS1 deletion was also detected and refined between exon 3 up to exon 11 (20 Kb) by target capture and sequencing (P4) (Figure 3). In addition, a novel mutation (c.779T>A, p.V260E) was identified in LIS1 in one of the boys (P5) and two other novel mutations (c.910G>C, p.G304R/ c.436T>C, p.F146L) were found in DCX in 2 boys (P7,P8) by target capture and sequencing (Table 1) (Figure 4). In fact, mutation in exon 8 of LIS1 gene was not found in any of the patient’s parents, confirming the de novo origin in the patient (P5) (Figure 4). The mutation c.910G>C of the DCX gene was validated by Sanger sequencing in the patient but in none of the parents indicating a de novo mutation (P7) (Figure 4). On the other hand, the mutation c.436T>C of the DCX gene was found in the patient’s mother (P8) (Figure 4).


In silico analyzes of the two variations (p.F146L) and (p.G304R) in the DCX gene showed that these two variations are predicted as “Disease Causing” (0.99) (Mutation Taster). Te variation (p.F146L) is possibly pathogenic with a score of 0.941 (polyphen-2). While the variation (p.G304R) is probably pathogenic with a score of 1 (polyphen-2). The F and G residue are highly preserved among different species (Phast Cons). Furthermore, in silico predictions demonstrated that the p.V260E variation in the LIS1 gene was likely predicted as deleterious ("Disease Causing" (0.99) (Mutation Taster)) and as probably pathogenic (polyphen-2/score=0,999). Furthermore, the V residue is highly preserved among different.
Discussion
In the past few years, genetic studies identified 20 genes associated with lissencephaly [11], including LIS1 (OMIM * 607432) and DCX (OMIM * 300121), encoded two proteins associated to microtubules and initially identified as the causative genes of this disorder. So far, mutations in these genes account for 85% of type 1 lissencephaly [12]. LIS1 mutation is mostly associated with type 1 lissencephaly whereas, hemizygous DCX mutation is less common and is typically observed in males [4].
On the other hand, subcortical band heterotopia is observed about 10 times more frequently in females [13]. Clearly, this is in accordance with our cohort of patient.
Target capture sequencing identified a novel heterozygous missense mutation of LIS1 (c.779T>A) in a male patient (P5), would result in a non-functional protein (p.V260E), which is located on the 5 WD40 repeats. Interestingly, the 260-valine acid residue is highly preserved among different species, indicating its importance to the protein function in the development of the nervous system. According to bioinformatic analyses, we suggest that this mutation is a novel mutation and is predicted to be potentially damaging. To date, most of the mutations reported in LIS1 gene are deletions, duplications and nonsense while missense mutation represents approximately 16% of all mutations reported [4]. Furthermore, missense mutations are associated with less severe phenotype (pachygyria/grade 4) than truncating mutations (agyria/grade 2,3) [14]. In concordance with the phenotype of our patient who present pachygyria, it has been supposed that this deleterious mutation is responsible for this phenotype. However, others studies not have shown a correlation between the mutation’s type and the phenotype’s severity. For these reason, functional studies are required.
We have diagnosed 4 cases of 17p13.3 deletions : 3 patients (P2, P3 and P4) are responsible for Miller-Dieker Syndrome (MDS : OMIM#247200) and 1 patient (P1) is responsible for Isolated Lissencephaly (ILS : OMIM#607432). All patients presented type 1 lissencephaly (Agyria-Pachygyria). According to various previous studies, our patients shared common clinical feature [15-16]. Curiously, it has been reported that 17p13.3 deletion is associated with epilepsy (71.3%), lissencephaly (41%), intrauterine growth retardation (62%), ventricular dilation (59%) and corpus callosum anomalies(17%) [17,18].
It has been shown that MDS is caused by deletion including at least PAFAH1B1 and YWHAE genes and that ILS is caused by deletion and heterozygous mutation of PAFAH1B1. MDS patients exhibit more severe phenotype compared to patients with ILS [8-18]. This is consonant with clinical data of patients (P1, P2, P3 and P4). It has been reported that 90% of MDS patients are associated with 17p13.3. Also, partial deletions of PAFAH1B1 gene are responsible for rare MDS cases [19]. The clinical picture of MDS patients reported with both 17p13.3 or PAFAH1B1 deletions is characterized by severe lissencephaly [19-8], ascertainning the agyria observed in the patient (P4). Furthermore, array CGH showed a gain of nearly 26 Kb of the 17p arm in this patient (P4), between 2522672 and 2549373 based on the human genome version 18. Subsequently, this deletion was confirmed and refined between exon 3 up to exon 11 by target capture sequencing. Thus, we can hypothesize that FISH, CGH and Next Generation Sequencing (NGS) are complementary techniques. Despite the development of NGS, such as target capture sequencing which may be a useful tool in screening a large number of known causative gene mutations, CGH still remains an important technique with a high sensitivity in the detection of LIS1/DCX deletion.
Several variations in the DCX gene have been reported as responsible for anterior lissencephaly and Subcortical Band Heterotopia (SBH), second form of lissencephaly, in both boys and girls, respectively, who present essentially epilepsy and intellectual disability. These variations including deletions, nonsense mutations, frameshift mutations and missense mutations are located mostly in the two functional microtubule binding domains of doublecortin (N-DC and C-DC). However, the majority of mutations are de novo and approximately 20% are mutations inherited from the mother.
In our study, we identified two mutations in the DCX gene (c.910G>C; p.G304R) de novo and (c.436T> C, p.F146L) inherited (maternal), respectively in two male patients (P7) and (P8). A novel hemizygous missense mutation (c.910G>C), would result in a non-functional protein (p.G304R), that is located next to the C-DC domain. Due to its importance in the stabilization of microtubules during neuronal migration, we can suggest that variation can be responsible for the phenotype of patient (P7) who presented mental retardation, epilepsy, hypotonia and lissencephaly.
The second patient (P8) presented with mental retardation, delayed walking and lissencephaly. Targeted panel was identified a novel hemizygous missense mutation (c.436T>C), would result in a non-functional (p.F146L), that is located in the N-DC domain of the doublecortin protein. The 146-F acid residue is highly preserved among different species, and the mutation was estimated in silico as deleterious with a score of 0.941.
The patient (P8) has a phenotype less severe than that presented by the patient (P7). Thus far, we can suggest that p.F146L mutation affecting the N-DC domain has a less deleterious effect, likewise predicted by bioinformatics analysis (score=0.941).While, previous studies hypothesized that mutations in the C-DC domain resulted in a less severe phenotype compared to those in the N-DC domain [21]. In fact, it has been shown that the first 213 amino acids of DCX are enough for binding microtubules [22]. Therefore, a mutation beyond that would be associated with a less severe phenotype. These is not the case for the patient (P8), who has p.F146L mutation associated with the less severe phenotype compared to that presented by the patient (P7) with a p.G304R mutation. This difference could be explain by the capacity of the residue in itself to alter the three-dimensional structure and the stability and the function of the protein. Thus, functional studies are required in order to comparing these two variations. Interestingly, previous studies have shown that doublecortin functions via binding to microtubules, enhancing microtubule polymerization and facilitating microtubule bundling [23], suggesting that both N-CD and C-DC domain play an important role in microtubule binding [24]. Consequently, these mutations found out in our study may affect tubulin binding through direct interaction or destabilization of DCX protein and may contribute to the patients’ phenotype.
Conclusion
This is the first study in Tunisia reporting the clinical and genetic data in eight children with type 1 lissencephaly. In this study, we confirm that LIS1 and DCX are the main causative genes for this neuronal migration disorder. So far, deletion or mutation of these genes plays an important role in lissencephaly, with each gene leading to a specific feature although genetic interactions between these genes also exist. Furthermore, our study contributes to a better delineation of the genotype-phenotype correlation of type 1 lissencephaly and spotlights the usefulness of developing approaches and methods for detecting a large number of known causative gene mutations.
Declarations
Ethics approval and consent to participate: This study was approved by the local Ethics Board of the University Teaching Hospital Farhat Hached. Written informed consent to participate in this study was obtained from the parents.
Consent for publication: Written informed consent was obtained from the parents for photo and clinical data publication.
Availability of data and materials: All data generated or studied during this study are included in the published article which is available upon request from the corresponding author.
Competing interests: All the authors have no competing interests.
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Authors’ contributions: Soumaya Mougou-Zerelli contributed to conception and design. Jamel Chelly, Meriam Hadj Amor, Wafa Slimani, Nathalie Drouot, Sarra Dimassi, Hanene Hannachi and Hela Ben Khelifa contributed to all experimental work, analysis and interpretation of data. Jamel Chelly and Nathalie Drouotcoordinated the molecular analysis and validated the results (NGS). Nejla Soyah, Adnene Mlika, Hedia Klai, Ichraf Kraoua, Ilhem Turki, Khaled Ben Helel, Naziha Gouider-khoujaand Lamia Boughamoura referred patients to our department. Soumaya Mougou-Zerelli, Sarra Dimassi and Ahlem Attig were responsible for the consultation. Soumaya Mougou-Zerelli and Ali Saad were responsible for overall supervision. Meriam Hadj Amor drafted the manuscript, which was revised by Soumaya Mougou-Zerelli. All authors read and approved the final manuscript.
Acknowledgements: We are very grateful to the family members for their kind participation and for their continuous interest in this study. We also thank the scientific and technical team of the cytogenetics Department at Farhat Hached University Teaching Hospital (Sousse, Tunisia) and Ms. N. Kerkni for English editing.
Supplementary information: Additional file 1: Figure S1. Pedigree of the family. The proband (gray) and her sister (striped) carried a der (3) and der (17) respectively. The white triangle and the black diamond represent terminated pregnancies and affected stillborn, respectively.
References
- Guerrini R, Dobyns W B. Malformations of cortical development: Clinical features and genetic causes. Lancet Neurol. 2014; 13: 710-726.
- Cardoso C, Leventer RJ, Matsumoto N, Kuc JA, Ramocki MB, Mewborn SK, et al. The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum Mol Genet. 2000; 9: 3019-3028.
- Mokánszki A, Körhegyi I, Szabó N, Bereg E, Gergev G, Balogh E, et al. Lissencephaly and Band Heterotopia: LIS1, TUBA1A, and DCX Mutations in Hungary. J Child Neurol. 2012; 27: 1534-1540.
- Uyanik G, Morris Rosendahl DJ, Stiegler J, Klapecki J, Gross C, Berman Y, et al. Location and type of mutation in the LIS1 gene do not predict phenotypic severity. Neurology. 2007; 69: 442- 447.
- Jamuar SS, Lam ATN, Kircher M, D’Gama AM, Wang J, Barry BJ, et al. Somatic mutations in cerebral cortical malformations. New England Journal of Medicine. 2014; 371: 733–743.
- Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw Boris A, et al. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian LIS1. Nat Cell Biol. 2000; 2: 767-775.
- Gressens P. Pathogenesis of migration disorders. Curr Opin Neurol. 2006; 19: 135-140
- Cardoso C, Leventer RJ, Ward HL, Toyo-oka K, Chung J, Gross A, et al. Refinement of a 400-kb Critical Region Allows Genotypic Differentiation between Isolated Lissencephaly, Miller-Dieker Syndrome, and Other Phenotypes Secondary to Deletions of 17p13.3. The American Journal of Human Genetics. 2003; 72: 918–930.
- Matsumoto N, Leventer RJ, Kuc JA, Mewborn SK, Dudlicek LL, Ramocki MB, et al. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur J Hum Genet. 2001; 9: 5–12.
- Mc Gowan-Jordan J, Simons A, Schmid M. An international system for human cytogenomic nomenclature. Cytogenet Genome Res. 2016; 149: 1–2.
- Di Donato N, Timms AE, Aldinger KA, Mirzaa GM, Bennett JT, Collins S, et al. Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet Med. 2018; 20: 1354- 1364.
- Pilz DT, Matsumoto N, Minnerath S, Mills P, Gleeson JG, Allen KM, et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Human Molecular Genetics. 1998; 7: 2029–2037.
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998; 92: 63–72.
- Leventer RJ, Cardoso C, Ledbetter DH, Dobyns WB. LIS1 missense mutations cause milder lissencephaly phenotypes including a child with normal IQ. Neurology. 2001; 57: 416–422.
- Blazejewski SM, Bennison SA, Smith TH, Toyo Oka K. Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3. Frontiers in Genetics. 2018; 9: 80.
- Parrini E, Conti V, Dobyns WB, Guerrini R. Genetic Basis of Brain Malformations. Mol Syndromol. 2016; 7: 220–233.
- Chen CP, Liu YP, Lin SP, Chen M, Tsai FJ, Chen YT, et al. Ventriculomegaly, Intrauterine Growth Restriction, and Congenital Heart Defects as Salient Prenatal Sonographic Findings of Miller-Dieker Lissencephaly Syndrome Associated With Monosomy 17p (17p13.2 → pter) in a Fetus. Taiwanese Journal of Obstetrics and Gynecology. 2010; 49: 81–86.
- Güngör S, Yalnizoǧlu D, Turanli G, Saatçi I, Erdoǧan Bakar E, Topçu M. Malformations of cortical development: Clinical spectrum in a series of 101 patients and review of the literature (Part I). Turkish Journal of Pediatrics. 2007; 49: 120–130.
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, et al. Isolation of a Miller Dicker lissencephaly gene containing G protein β-subunit-like repeats. Nature. 1993; 364: 717–721.
- Izumi K, Kuratsuji G, Ikeda K, Takahashi T, Kosaki K, et al. Partial Deletion of LIS1: A Pitfall in Molecular Diagnosis of Miller Dieker Syndrome. Pediatric Neurology. 2007; 36: 258–260.
- Leger PL, Souville I, Boddaert N, Elie C, Pinard JM, Plouin P, et al. The location of DCX mutations predicts malformation severity in X-linked lissencephaly. Neurogenetics. 2008; 9: 277-285.
- Horesh D, Sapir T, Francis F, Wolf SG, Caspi M, Elbaum M, et al. Doublecortin, a stabilizer of microtubules. Human Molecular Genetics. 1999; 8: 1599–1610.
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999; 23: 257-271.
- Moreira I, Bastos Ferreira R, Silva J, Ribeiro C, Alonso I, Chaves J, et al. Paternal transmission of subcortical band heterotopia through DCX somatic mosaicism. Seizure. 2015; 25: 62-64.