
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Series - Open Access, Volume 3
Fabiana Vercellino
Nikita Mehtani*; Gerard Thong; Chadwan Al-Yaghchi; Gurpreet Sandhu
Child Neuropsychiatry Unit, SS. Antonio e Biagio e Cesare Arrigo Hospital, Spalto Marengo 46, 15121, Alessandria, Italy
*Corresponding Author: Fabiana Vercellino
Child Neuropsychiatry Unit, SS. Antonio e Biagio e
Cesare Arrigo Hospital, Spalto Marengo 46, 15121,
Alessandria, Italy
Email: fvercellino@ospedale.al.it
Received : Jan 02, 2022
Accepted : Feb 14, 2022
Published : Feb 21, 2022
Archived : www.jcimcr.org
Copyright : © Vercellino F (2022).
Citation: Vercellino F. Brain MR imaging findings in children with congenital muscular dystrophies. J Clin Images Med Case Rep. 2022; 3(2): 1678.
Introduction
Congenital muscular dystrophies (CMDs) are a heterogeneous group of disorders presenting early in life during infancy or soon after birth with muscle weakness and hypotonia, sometime associated to severe brain involvement and histologically presenting with dystrophic lesions. They are classified on the basis of the clinical features, pathologic findings and pattern of inheritance. In fact most of these disorders are inherited and linked to specific genes. Several CDMs classifications have been proposed [1-3]. The main CMD subtypes, classified by pathogenic gene, are laminin alpha‐2 (merosin) deficiency (MDC1A), collagen VI-related CMD (COL6-RD), the dystroglycanopathies -such as Walker‐Warburg syndrome (WWS), Fukuyama CMD (FCMD), and muscle-eye-brain disease (MEB)-, SELENON (SEPN1)‐related CMD, and LMNA‐related CMD (L‐CMD) [4]. The incidence and prevalence of CMDs in various populations is not sufficiently known and may have been underestimated in early published CMD surveys owing to more limited diagnostic means available. Point prevalence in various studies ranges from 0.56 to 2.5 per 100,000 [5-10]. The relative frequency of CMD subtypes also varies in different populations. The most common CMD subtypes are laminin-α2 related CMD and dystroglycanopathies, followed by collagen VI-related CMD. The forms of congenital muscular dystrophy related to mutations in SEPN1 and LMNA are less frequent [9,11]. CMDs are often autosomal recessive, but some cases have been found to follow autosomal dominant patterns, by direct inheritance, spontaneous mutations, or mosaicism. Clinical genetic testing is available for virtually all genes known to be associated with CMDs. However, many affected individuals remain without a genetic diagnosis, an indicator that novel disease genes have yet to be identified [3]. Differential diagnosis of patients presenting with weakness in early infancy includes: Congenital myopathies like central core, nemaline road, and centronuclear myopathy and those secondary to metabolic disorders; Disorders of the myoneural junction including congenital myasthenia gravis and infant botulism and Neuropathies like spinal muscular atrophy (SMA) and hereditary motor sensory neuropathy (HMSN) [12]. Nowadays the diagnosis is based on clinical presentation, laboratory tests and genetic investigations. Muscular biopsy can be an important diagnostic tool if genetic tests are not available. Brain MRI and muscular MRI are performed if available. We discuss brain MR imaging findings in our patients with CMDs trying to outline common features and differences in order to aid in the diagnosis of these rare disorders before performing genetic tests.
Materials and methods
Four children with CMDs were retrospectively analyzed: n.1 with merosin-deficient CMD (MDC1A); n.2 and n.3 with Walker Warburg syndrome (WWS); n.4 with Ullrich myopathy (COL6-RD). Two were boys and two were girls. Case n.2 and n.3 were siblings. Parental consanguinity was found in all the families. One family was Turkish, the other two were Moroccan. All the patients showed general hypotonia and muscle weakness at birth. In patients with merosin-deficient CMD and Walker Warburg myopathy phosphokinase (CPK) concentration was markedly elevated (>1000 U/l, normal range<175 U/l), while was only mildly elevated in patient with COL6-RD. Genetic tests were performed in two of the four patients, one patient (n.2) was dead at the time of the study, her parents refused genetic tests for their daughter still alive. Children were studied with MRI of the brain. MRI were obtained on a 1.5-T imager. T1-weighted sagittal and axial images, T2-weighted sagittal and coronal images were obtained in all four patients. The MRI studies were analyzed for structural abnormalities of the cerebrum and cerebellum, cortical migration anomalies and white matter disorders. We also determined the presence of enlarged subarachnoid spaces, ventriculomegaly, abnormalities of the brain stem.
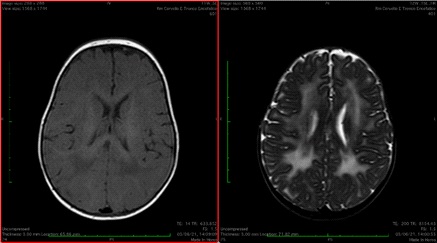
Patient 1 was the third child of a Moroccan couple. There was a history of consanguinity in parents (first cousins). Pregnancy and delivery were uneventful. At birth he had severe hypotonia and muscle weakness but he did not require ventilatory assistance. His head circumference was normal, no dysmorphic features were present. At the age of nine months (last follow up) he was still unable to hold his head up. He did not have respiratory problem. He was able to eat by mouth but his weight gain was poor. On examination he had myopathic facies and pectus carinatum. He was hypotonic with absent deep tendon reflexes and joint contractures of distal lower extremities. Speech development was normal (babbling). At birth his serum CPK was 46540 U/l. His echocardiogram was normal. EEG was unremarkable. We analyzed the LAMA2 gene sequencing and revealed a homozygous mutation c.3976C>T. The same mutation was found in both the parents in heterozygosis. Due to the result of genetic test, muscle biopsy was not performed. Magnetic resonance imaging (MRI) of the brain, performed when he was 5 months, showed diffuse high signal in the periventricular and subcortical white matter especially in the parieto-occipital lobes (Figure 1).
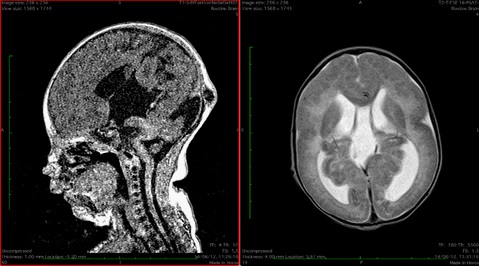
The patient 2 and 3 were sisters. Their parents were consanguineous (first cousins), the family came from Morocco. The patient 2 was born at term. During pregnancy a severe cerebral malformation was revealed by prenatal ultrasound. From birth she exhibited marked hypotonia with generalized muscle weakness and respiratory difficulties. CPK was elevated (1100 U/l). She underwent a brain MRI which revealed cortical dysplasia (type II lissencephaly, Cobblestone type), multiple heterotopic subependymal nodules, mid brain kinking, cerebellum hypoplasia, high signal in the periventricular and subcortical white matter, hydrocephalus, occipital meningocele (Figure 2). Echocardiography was normal. She was unable to breath without support. She was unable to suck so a nasogastric tube for feeding was necessary. On examination the patient was hypotonic, deep tendon reflexes were diminished. She had joint contractures of upper and lower extremities, myopathic facies, microphthalmia. Her head circumference was normal at birth but increased due to hydrocephalus. Parents did not give consent for ventriculoperitoneal shunt. In the following years her motor milestones were markedly delayed. She was unable to hold her head up and to sit alone. Language was absent. She died when she was five years old. Clinical and radiological aspects were suitable for Walker Warburg syndrome.
The patient 3 was the sister of patient 2. Her problems were the same. She was born at term, by caesarian section performed for cerebral malformation revealed by prenatal ultrasound. She had global hypotonia, generalized muscle weakness and respiratory difficulties. Her head circumference was normal at birth but increased due to hydrocephalus. She showed two small occipital and parietal meningoceles. Her serum CPK level was 4370 U/l. EEG showed hypostructured activity with bilateral slow waves. Cerebral MRI revealed cortical dysplasia (type II lissencephaly, Cobblestone type) over temporo-occipital areas, cerebellar hypoplasia especially of the vermis, mid brain kinking, small posterior fossa, high signal in the periventricular and subcortical white matter. Triventricular hydrocephalus with partial pellucid sept agenesis and hypoplasic optic bulbs with abnormalities of the crystalline and vitreous, retinal detachment were also present (Figure 3). Clinical and radiological aspects were suitable for Walker Warburg syndrome. The head circumference increased since the first days of life and a ventriculoperitoneal shunt was necessary. At the last visit the child was 12 months. Her motor milestones were markedly delayed. She was unable to hold her head up and to sit alone. Language was absent. She was able to breath without support but she was fed by nasogastric tube. Clinical and radiological aspects were suitable for Walker Warburg syndrome.The parents refused to do genetic tests for both.
Patient 4 was the fifth child of healthy consanguineous (first cousins) Moroccan parents. His head circumference was normal. Global hypotonia, proximal elbow and knee contractures and hyperlaxity of the distal joints were noted at birth. He had congenital hip dislocation. He acquired ambulation, showing a mild delay (18 months). At last follow up clinical examination showed bilateral proximal muscle weakness, affecting the upper more than the lower limbs, distal laxity in the wrist and in the extensor of the fingers, proximal contractures, spinal involvement with kyphosis and severe scoliosis, talipes equinovarus. Muscle weakness was stable up to 5 years; then progressively worsened with increasing age, resulting in frequent falls, difficulty in standing up and raising arms above the shoulders. He presented severe tendon Achilles tightness, corrected by surgical lengthening at 6 years old; after surgery, he didn’t restart walking. Cognitive, language and social development were appropriate. Examinations of the respiratory systems (spirometry) was normal; echocardiography and serial EKG were normal. Serum creatine kinase (CK) levels was only mildly elevated (max 493 U/l). Other hematological test results were normal. Electromyography (EMG) presented mild myopathic abnormalities and motor and sensory nerve conduction study was normal. The muscle biopsy (H&E stain, modified Gömöri Trichrome, oxidative stains, ATPase stains) showed the typical histological pattern of muscle anomalies of dystrophic lesions. Immunohistochemical examination showed a complete absence of collagen VI immunoreactivity. Genetic tests found an homozygous deletion c.6284delG (p. Gly2095Alafs*12) in exon 18 of the COL6A3 coding sequence (encoding the α3 chains of collagen VI) carrying a pathogenetic significance. Brain MRI findings were normal.
Results
Case summaries for all four children are presented in Table 1 and 2.
Discussion
Congenital muscular dystrophies (CMDs) are genetically and clinically heterogeneous hereditary myopathies. Congenital hypotonia and muscular weakness, decreased or absent deep tendon reflexes, delayed motor milestones, CPK elevation and white matter and grey disorders on brain MR are common features.
Usually patients with merosin-deficient CMD have normal head circumference, normal or near-normal intelligence, and high CPK levels. Contractures and joint deformities may be present at birth [13]. Philpot et al [14] collected clinical features and brain imaging in 24 cases of CMD in relation to the merosin status. Magnetic resonance imaging of the brain was carried out on 15 of the children. All eight merosin-positive cases had normal scans whereas all seven of the merosin-deficient cases had significant changes in the white matter. This study has demonstrated that children with merosin-deficient CMD have a more severe clinical phenotype and associated white matter changes on brain imaging. Leite et al [15] evaluated 25 patients with partial or total merosin deficiency using MRI. Bilateral WM involvement was seen to be more prominent in the parietal, frontal, and temporal regions of the brain. The brain stem and internal and external capsules were less affected. Cerebellar WM involvement was rare. This series of patients demonstrated that there was no correlation between the extent of WM abnormality on MRI and the clinical status and degree of merosin deficiency (partial or total). Changes on follow-up imaging studies did not correlate with the clinical status of the patient. Di Blasi et al [16] revealed white matter changes in all cases on MRI performed in 8 patients with merosin-negative CMD. Oliveira et al [17] performed MRI in 22/26 patients with merosin-negative CMD and revealed white matter changes in all cases. In addition, they detected other cerebral changes included abnormal gyration in three patients. In patients with merosin-deficient CMD Caro et al [18] suggested that the increased T2 prolungation time on brain MR may be caused by increased water content in the white matter due to an abnormal blood-brain barrier rather than decreased or abnormal myelination. This finding could be consistent with the normal or near-normal intelligence seen in merosin-deficient CMD while leukodystrophies are usually associated with symptoms due to CNS involvement.
Clinical presentation of dystroglycanopathies is heterogeneous ranging from early and severe clinical involvements, as demonstrated by the WWS, MEB, and FCMD, to later and less pronounced muscle impairment and with/without comorbidity with other body-organs [19-21]. The most severe forms are associated with head circumference above the average, mental retardation and high CPK levels [22]. Fukuyama-type congenital muscular dystrophy (FCMD), muscle-eye-brain disease (MEB), and Walker-Warburg syndrome (WWS) are autosomal recessive disorders characterized by congenital muscular dystrophy with structural brain and eye abnormalities. Initially, alpha-DGP was classified under congenital muscular dystrophies; however, the clinical phenotype is now expanded to include a markedly wide spectrum ranging from the most severe, lethal congenital muscular dystrophy with severe brain deformity to the mildest limb girdle muscular dystrophy with minimal muscle weakness. As stressed by Martin [23] mutations in 6 genes are now known to give rise to forms of congenital or limb-girdle muscular dystrophy where defects in O-linked glycosylation are responsible, the so-called dystroglycanopathies. Genes for Walker Warburg syndrome, muscle-eye-brain disease, Fukuyama congenital muscular dystrophy, congenital muscular dystrophy 1C and 1D, and limb girdle muscular dystrophy 2I have been identified. The notion that each disorder is clinically distinct and is caused by a mutation in a distinct gene, a theme prevalent in the early literature, is incorrect. Some generalities, however, remain. For example, WWS, MEB, and FCMD all share common brain malformations in addition to muscular dystrophy, whereas brain malformations are far less common in MDC1C and are not present in LGMD2I. The most common brain finding in WWS, FCMD, and MEB is type II (cobblestone) lissencephaly. In WWS, the brain can be almost completely agyric. Other brain findings can include dilation of the cerebral ventricles, flattened brainstem, absent corpus collosum, aberrant myelination, and occasional occipital encephalocele. As stressed by Bonneman et al [24] the hallmark of central nervous system involvement in the dystroglycanopathies, on brain MRI is represented by the cobblestone complex, ranging from complete lissencephaly (type II) to more focal pachygyria or polymicrogyria showing a frontal predominance. Typical findings on brain MRI include high signal in the white matter on T2 weighted and FLAIR images and are seen in all patients but are most obvious in patients greater than 6 months of age. The internal capsule, corpus callosum, and other dense fiber tracts are usually spared, but there may be subcortical cyst formation. White matter abnormalities on MRI are also seen in patients with incomplete deficiency, while patients with very late adulthood onset may have normal brain MRI.
Ullrich CMD typically presents in the newborn period with striking distal joint hypermobility of the hands and often feet with prominent calcanei, while talipes equinovarus can also occur. Congenital hip dislocation is frequently present. Proximal elbow and knee contractures, kyphoscoliosis, and torticollis may be also present at birth and may improve initially with physical therapy and orthopedic treatment. Cognition is normal and often advanced for age. CK is normal or mildly elevated. Neonatal respiratory failure, severe feeding difficulties, congenital contractures or major joint hyperlaxity would be highly unusual presenting features. Muscle biopsy showed dystrophic changes with absent staining for collagen. Mutations in one of the three COL6 alpha genes (COL6A1, COL6A2, and COL6A3) can lead to the COL6-RD spectrum, ranging from early onset, severe Ullrich CMD (UCMD) to an intermediate severity phenotype to milder Bethlem myopathy (BM) [25]. Brain MR is not usually performed in Ullrich CMD patients.
In our series MRI findings concerning white matter were strikingly similar for three out of four patients. On T2-weighted images three cases had a diffuse and symmetrical increase in signal in the white matter of the cerebral hemispheres. Type II (cobblestone) lissencephaly, mid brain kinking, cerebellum hypoplasia, hydrocephalus and occipital meningocele were present in patient 2 and 3 as typically in WWS. The brain stem and the cerebellum were structurally normal in patient 1 and 4. Our experience is similar to that found in the literature [18] in that CNS involvement with MR white matter abnormalities could be present without clinical evidence (see patient 1 with normal cognition). As stressed by Kang et al [3], many studies demonstrated that abnormal findings on brain imaging studies can predict the subtype-specific diagnosis in some cases, especially in merosinopathy (white matter abnormalities) and in some dystroglycanopathies (polymicrogyria, white matter lesions, pontine hypoplasia, and subcortical cerebellar cysts).
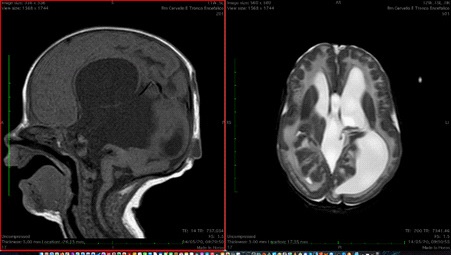
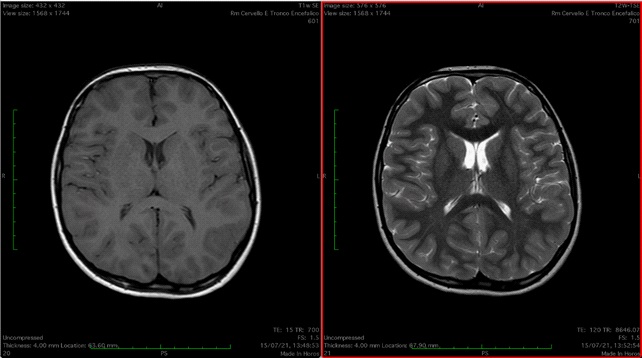
Table 1:Case summaries.
Patient |
Age at |
Clinical symptoms at birth |
CPK (U/l) |
Genetic diagnosis |
1 |
2 months |
hypotonia and muscle weakness; joint contractures |
46540 U/l |
LAMA2 gene homozygous mutation c.3976C>T |
2 |
1 month |
hypotonia and muscle weakness; feeding and respiratory problems |
1100 U/l |
no |
3 |
1 month |
hypotonia and muscle weakness; feeding and respiratory problems |
4370 U/l |
no |
4 |
8 months |
hypotonia and hyperlaxity of the distal joints |
493 U/l |
COL6A3 gene homozygous deletion c.6284delG |
CPK: creatine phosphokinase. No: not performed
Table 2: Brain MRI findings.
lissencephaly with cobblestone cortex |
cerebellar hypoplasia |
mid brain kinking |
No |
Yes |
No |
small posterior fossa, partial pellucid sept agenesis, encephaloceles |
||
4 |
No |
No |
No |
No |
No |
No |
No |
No |
MRI: magnetic resonance imaging; WM: white matter
Conclusion
Several conditions may manifest with infantile hypotonia, including anomalies of the central or peripheral nervous system with involvement of the spinal cord, anterior horn cell, peripheral nerves, neuromuscular junction, and muscles. Different muscular diseases share common clinical symptoms such as hypotonia and weakness, contractures, delayed motor milestones. The diagnostic evaluation is not easy mainly in differentiating the various types of CMDs, and represents a challenge for the neonatologists and pediatricians. Brain MRI findings play an important role in suspecting a specific CMD subtype in order to prioritize testing to arrive at a final genetic diagnosis. Brain MR imaging findings may help the clinicians in the diagnosis of rare disorders before performing genetic tests. Performing clinical and molecular diagnosis is extremely important for genetic counseling, prognosis, and anticipatory or prospective treatment. Once the pathogenic variant(s) have been identified in an affected family member, it is possible to perform prenatal testing for a pregnancy at increased risk and preimplantation genetic diagnosis for CMD.
References
- Bönnemann CG. The collagen VI‐related myopathies: Muscle meets its matrix. Nat Rev Neurol. 2011; 7: 379‐90. doi: 10.1038/ nrneurol. 2011. 81.
- Bönnemann CG, Wang CH, Quijano-Roy S, Deconinck N, Bertini E, Ferreiro A, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014; 24: 289-311. doi:10.1016/j.nmd.2013.12.011.
- Caro P A, Scavina M, Hoffman E, Pegoraro E, Marks HG. MR imaging findings in children with merosin-deficient congenital muscular dystrophy. Am J Neuroradiol 1999; 20: 324-326.
- Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord. 2000; 10: 1-9. doi: 10.1016/s0960- 8966(99)00055-3.
- Di Blasi C, Bellafiore E, Salih MA, Manzini MC, Moore SA, Seidahmed MZ, et al. Variable disease severity in Saudi Arabian and Sudanese families with c.3924 + 2 T > C mutation of LAMA2. BMC Res Notes. 2011; 4: 534. doi: 10.1186/1756-0500-4-534.
- Falsaperla R, Praticò AD, Ruggieri M, Parano E, Rizzo R, Corsello G, et al. Congenital muscular dystrophy: from muscle to brain. Ital J Pediatr. 2016; 42: 78. doi: 10.1186/s13052-016-0289-9.
- Fu XN, Xiong H. Genetic and Clinical Advances of Congenital Muscular Dystrophy. Chin Med J. 2017; 130: 2624-31. doi: 10.4103/0366-6999.217091.
- Graziano A, Bianco F, D’Amico A, Moroni I, Messina S, Bruno C, et al. Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology. 2015; 84: 904-11. doi: 10.1212/ WNL.0000000000001303.
- Hughes MI, Hicks EM, Nevin NC, Patterson VH. The prevalence of inherited neuromuscular disease in Northern Ireland. Neuromuscul Disord. 1996; 6: 69-73. doi: 10.1016/0960- 8966(94)00017-4.
- Incecik F, Herguner OM, Ceylaner S, Altunbasak S. Merosin-negative congenital muscular dystrophy: Report of five cases. J Pediatr Neurosci. 2015; 10: 346-9. doi: 10.4103/1817-1745.174432.
- Kang PB, Morrison L, Iannaccone ST, Graham RJ, Bönnemann CG, Rutkowski A, Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015; 84: 1369- 78. doi: 10.1212/WNL.0000000000001416.s
- Leite CC, Lucato LT, Martin MG, Ferreira LG, Resende MB, Carvalho MS, et al Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol. 2005; 35: 572-9. doi: 10.1007/s00247-004-1398-y.
- Mah JK, Korngut L, Fiest KM, Dykeman J, Day LJ, Pringsheim T, et al Systematic Review and Meta-analysis on the Epidemiology of the Muscular Dystrophies. Can J Neurol Sci. 2016; 43: 163-77. doi: 10.1017/cjn.2015.311.
- Martin PT. The dystroglycanopathies: the new disorders of Olinked glycosylation. Semin Pediatr Neurol. 2005; 12: 152-8. doi: 10.1016/j.spen.2005.10.003.
- Mostacciuolo ML, Miorin M, Martinello F, Angelini C, Perini P, Trevisan CP. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet. 1996; 97: 277-279. doi: 10.1007/BF02185752.
- Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy. Lancet. 2002; 360: 1419-21. doi: 10.1016/S0140-6736(02)11397-3.
- Muntoni F, Valero de Bernabe B, Bittner R, Blake D, van Bokhoven H, Brockington M, et al. 114th ENMC International Workshop on Congenital Muscular Dystrophy (CMD) 17-19 January 2003, Naarden, The Netherlands: (8th Workshop of the International Consortium on CMD; 3rd Workshop of the MYO-CLUSTER project GENRE). Neuromuscul Disord. 2003; 13: 579-88. doi: 10.1016/S0140-6736(02)11397-3.
- Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: indepth analysis of a muscle clinic population. Brain. 2009; 132: 3175-3186. doi: 10.1093/brain/awp236.
- Oliveira J, Santos R, Soares-Silva I, Jorge P, Vieira E, Oliveira ME, et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. 2008;74:502-12. doi: 10.1111/j.1399-0004.2008.01068.x.
- Parano E, Fiumara A, Falsperla R, Vita G, Trifiletti RR. Congenital muscular dystrophy: correlation of muscle biopsy and clinical features. Pediatr Neurol. 1994; 10: 233-6. doi: 10.1016/0887- 8994(94)90029-9.
- Parano E, Pavone L, Fiumara A, Falsaperla R, Trifiletti RR, Dobyns WB. Congenital muscular dystrophies: clinical review and proposed classification. Pediatr Neurol. 1995; 13: 97-103. doi: 10.1016/0887-8994(95)00148-9.
- Parija D, Tadi P. Congenital Muscular Dystrophy. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan 31.
- Philpot J, Sewry C, Pennock J, Dubowitz V. Clinical phenotype in congenital muscular dystrophy: correlation with expression of merosin in skeletal muscle. Neuromuscul Disord. 1995; 5: 301-5. doi: 10.1016/0960-8966(94)00069-l.
- Reed UC. Congenital muscular dystrophy. Part I: a review of phenotypical and diagnostic aspects. Arq Neuropsiquiatr. 2009; 67: 144-68. doi: 10.1590/s0004-282x2009000100038.
- Sframeli M, Sarkozy A, Bertoli M, Astrea G, Hudson J, Scoto M, et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord. 2017; 27: 793-803. doi: 10.1016/j.nmd.2017.06.008.