
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Sacrococcygeal teratoma in Nigerians: A case report and clinico-pathologic review
Ukachukwu AK1,2*; Aghahowa ME3,4; Ezike KN5,6; NwokorieRM7; Nwaribe EE8; Anaeto SI1
1 Neurosurgery Unit, Department of Surgery, Asokoro District Hospital, Abuja, Nigeria.
2 Duke Global Neurosurgery and Neurology, Durham, North Carolina, USA.
3 General Surgery Unit, Department of Surgery, Asokoro District Hospital, Abuja, Nigeria.
4 Department of Surgery, College of Health Sciences, Nile University of Nigeria, Abuja, Nigeria.
5 Department of Pathology, Asokoro District Hospital, Abuja, Nigeria.
6 Department of Anatomic Pathology, College of Health Sciences, Nile University of Nigeria, Abuja, Nigeria.
7 Department of Anesthesia, Asokoro District Hospital, Asokoro, Abuja, Nigeria.
8 Neurosurgery Unit, Department of Surgery, National Hospital, Abuja, Nigeria.
*Corresponding Author: Alvan Ukachukwu
Consultant Neurosurgeon, Asokoro District Hospital,
PMB 203, Asokoro 900231, Abuja - FCT, Nigeria.
Email: alvan.ukachukwu@gmail.com;
dr_alvan@yahoo.com
Received : Dec 27, 2021
Accepted : Feb 16, 2022
Published : Feb 23, 2022
Archived : www.jcimcr.org
Copyright : © Ukachukwu A (2022).
Abstract
Introduction: Sacrococcygeal Teratomas (SCTs) are rare childhood tumors, commonly seen in the neonatal period. The mainstay of their diagnosis is both clinical and radiological. Early gross-total surgical excision has the best prognostic outcome, especially for benign tumors.
Case report: We present the case of a 12-year old girl with recurrent infected right gluteal benign SCT who was successfully managed in a secondary-level health facility in Nigeria. She has remained symptom-free with no recurrence four years post-surgery. We also reviewed the medical literature on SCT in Nigeria.
Discussion: Our case report and others from Nigeria are consistent with the global trend on SCTs. They mostly occur within the first 15 years of life, with a bimodal peak in the first 4 years and at 8-11 years. 75-90% are benign, and the tumors often contain tissues from the three germ layers. Complete surgical excision is the treatment of choice.
Conclusion: SCTs are rare tumors, but require early diagnosis and complete surgical excision to prevent malignant transformation. The case report draws attention to the need for a multi-institutional tumor registry in Nigeria and other sub-Saharan African countries.
Keywords: sacrococcygeal teratoma; surgical excision; nigeria; literature review.
Abbreviations: CT: Computed Tomography; MRI: Magnetic Resonance Imaging; SCT: Sacrococcygeal Teratoma.
Citation: Ukachukwu AK, Aghahowa ME, Ezike KN, Nwokorie RM, Nwaribe EE, et al. Sacrococcygeal teratoma in Nigerians: A case report and clinico-pathologic review. J Clin Images Med Case Rep. 2022; 3(2): 1686.
Introduction
Teratomas are tumors that arise from the proliferation of pluripotent stem cells which can differentiate into ectodermal, endodermal, and mesodermal derivatives in embryonic life [1,2]. These tumors occur in various gonadal and extragonadal regions, with Sacrococcygeal Teratomas (SCTs) being the most common extragonadal teratoma [2,3]. SCTs are rare childhood tumors [1-4]. They are, however, the most common tumor in the neonatal period [5-7]. SCTs account for up to 30% of all childhood teratomas [1] and usually present as benign or malignant forms. SCTs are very rare in adults, with less than 100 cases reported in Teratomas are tumors that arise from the proliferation of pluripotent stem cells which can differentiate into ectodermal, endodermal, and mesodermal derivatives in embryonic life [1,2]. These tumors occur in various gonadal and extragonadal regions, with Sacrococcygeal Teratomas (SCTs) being the most common extragonadal teratoma [2,3]. SCTs are rare childhood tumors [1-4]. They are, however, the most common tumor in the neonatal period [5-7]. SCTs account for up to 30% of all childhood teratomas [1] and usually present as benign or malignant forms. SCTs are very rare in adults, with less than 100 cases reported in the literature [8].
Several theories have been adduced to the origin of SCTs. The Budde-Willis hypothesis suggests that teratomas arise from cells released from the normal developmental control of the primary embryonic organizer [1]. The germ cell theory postulates an origin from primordial germ cells either within the gonadal anlage or in extragonadal sites following arrest in germ cell migration from the yolk sac to the gonadal anlage in embryonic life [1,8]. Other theories suggest derivation from multipotent cells in Henson’s node which migrate caudally to the coccygeal region; wandering germ cells derived from a non-parthenogenetic source, abandoned during the migration of embryonic germ cells from the yolk sac to the gonad; and origin in totipotential or pluripotential embryonic cells [3,8,9].
We report the successful management of a huge infected recurrent sacrococcygeal teratoma in a pre-teen Nigerian girl.
Case report
A 12-year-old girl presented with a huge lump in the right gluteal region from birth. Surgery was done at 3 months of age in a rural health center, but it soon recurred and progressively increased in size. There was associated recurrent, intermittent, occasionally colicky pain that was relieved with simple analgesics in the last 5 years which progressively increased in its frequency and duration, and was worse on defecation. There was significant wasting of the right lower limb, with associated weakness, tingling, and numbness. She had recurrent difficulty with micturition and defecation. The girl was the 8th of 15 children in a polygamous home. She had been living in an internally displaced persons camp for 3 years following the destruction of her community by the Boko Haram insurgents. The cost of her care was covered by donations from a philanthropic religious organization and waiver of some fees by the hospital management.
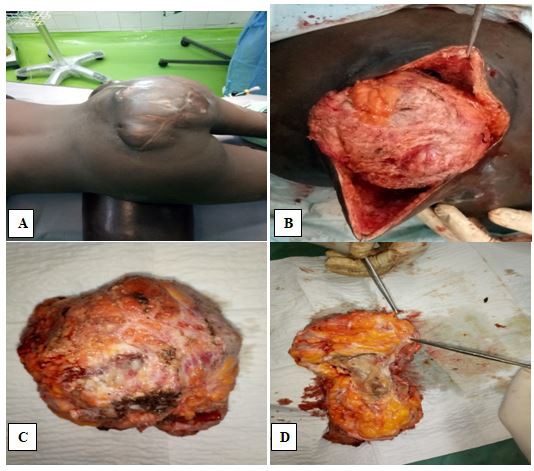
She was ambulant with the aid of walking crutches and had a large multilobulated mass measuring 13 X 12 X 11 cm in the right lower lumbar and sacral regions, extending across the midline to the left, with an irregular oblique hypertrophic scar over its mid aspect. The mass was soft and slightly tender, with hyperpigmented skin having multiple draining sinuses. The patient also had significant atrophy of the right lower limb with reduced muscle power (grade 3 on the right, grade 5 on the left) and hypoesthesia. Our clinical diagnosis was an infected sacrococcygeal teratoma.
Plain lumbosacral spine radiography did not show any abnormality. Plain pelvic radiography showed a right-sided hemipelvic soft tissue mass, with pelvic asymmetry evidenced by a small right hip and femur. Magnetic Resonance Imaging (MRI) of the lumbosacral spine (Figure 1) showed a well-defined oval-shaped sacrococcygeal mass measuring 12.4 X 13.5 X 12.7 cm, extending from the right lower lumbar region, across the midline, to the S3 presacral space. The mass was heterogeneous on T1W, T1 contrast, and T2W sequences, with multiple hypointense non-enhancing regions, divided by enhancing septations, suggesting areas of necrosis or loculated abscess collections. There was associated elevation/splaying of the adjacent sacrococcygeal bone, with mild straightening (but preserved alignment) of the lumbar spine. The other visualized vertebrae and intervertebral discs were normal in morphology and signal intensity. The urinary bladder had significant residual urine volume.

She was worked up for surgery and had pre-operative bowel preparation, with additional low rectal washout with soap and water enema on the morning of surgery. Intra-operatively, she received antibiotic cover and had gross-total en-bloc excision of the mass via a sacral approach done by combined neurosurgery and general surgery team. The intra-operative findings included an oblique hypertrophic scar over the mass and gluteal region, multiple discharging sinuses, and well-circumscribed encapsulated thick-walled mass with cystic portions containing purulent fluid and solid portions having bone and cartilaginous tissue in situ (Figure 2). There was no bowel involvement.
Postoperatively, she received culture-sensitive parenteral antibiotics for 5 days, and subsequent oral antibiotics for 14 days. She had transient urinary and fecal incontinence which resolved within 3 days, and superficial wound dehiscence which was managed with topical honey-impregnated dressings. The patient was discharged home on the 24th postoperative day for subsequent outpatient follow-up. She has remained symptomfree with no recurrence for four years.
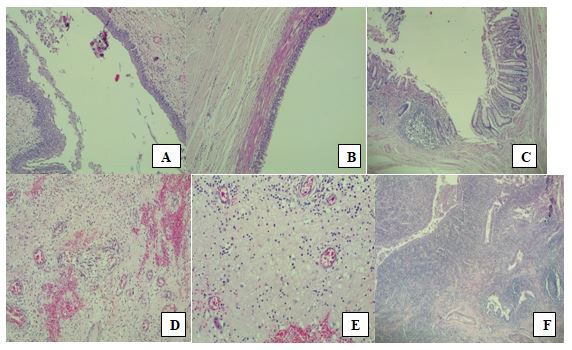
Macroscopically, the mass was 12 X 12 X 11 cm weighing 649g, with 2 accompanying skin tissue flaps measuring 14 X 12 cm and weighing 7 g each. Cut section through the mass showed it to be partly solid and partly cystic, and greyish-yellow in color. The cystic cavities were filled with whitish gelatinous materials. Microscopic sections (Figure 3) showed tissue covered by skin which exhibited hyperkeratosis and acanthosis of the epidermis which overlay a germ cell neoplasm composed of cystic structures and tissues from the 3 germ layers: mature skin with adnexal structures from ectoderm; pseudostratified ciliated respiratory epithelium and intestinal-type epithelium with goblet cells from endoderm; and immature cartilage from mesoderm. The stroma was densely infiltrated by mixed inflammatory cells including neutrophils, lymphocytes, plasma cells, and macrophages. Multiple foci of abscess collection were also seen. The histological diagnosis was Grade 1 Immature Teratoma.
Discussion
There is an increased interest in SCTs because of a rising hospital incidence [3,10]; estimated at 1 in 35,000 – 40,000 live births [4,6,8]. SCTs occur in patients between 1 day to 15 years of age in most case series [1-4], with a bimodal peak in the first 4 years of life and between 8 and 11 years [1,2]. A retrospective review of 74 cases from the pathology specimen register in Ibadan, South-West Nigeria, showed an average age of 4.3 years for all teratomas, with a range of 1 day to 15 years [1,2]. Our index patient (a 12-year old girl) falls within this age range. SCTs are, however, rare in Nigerian adults, as only a few reports document their presence in the adult population [8,12-14]. All the reports note that teratomas, including SCTs, are more common in females, with a female to male ratio of 5-18:1 [1-5,8,15].
Pathologically, SCTs have been classified as benign or malignant; cystic, solid, or mixed; and mature, immature, or malignant [1-3]. 75-90% of SCTs are benign, and males account for more of the malignant cases [7]. The malignant types have been further classified as embryonal carcinomas or yolk sac tumors, including endodermal sinus tumors [1-3]. Our patient had a benign mixed immature teratoma, similar to other Nigerian reports [1,2,5,12,13,16].
SCTs contain a variety of tissue types derived from the 3 germ cell layers – ectoderm, mesoderm, and endoderm [1]. Our patient’s histology showed tissue from all 3 germ layers: mature skin with adnexal structures from ectoderm; pseudostratified ciliated respiratory epithelium and intestinal-type epithelium with goblet cells from endoderm; and immature cartilage from mesoderm. A case report from Enugu, South-East Nigeria, noted the tumor contents to be mature adipocytes, skin and neural tissue [7], while another case series reported the presence of cardiac muscle in some of their excised SCT specimens [9]. In the pathology specimen series from Ibadan, derivatives of all 3 germ layers were seen, including breast tissue, salivary gland, hair, sebaceous material, and pancreatic tissue showing both islets of Langerhans and exocrine acini [1,2].
The clinical presentation of SCTs includes gluteal or sacral mass, sacrococcygeal pain, bladder dysfunction/acute urinary retention, and chronic constipation or fecal obstruction [3,4,8]. Other rare modes of presentation are neurologic symptoms (including intractable sciatica, loss of anal reflex, and lower limb weakness), abdominal and perineal pain, anemia, elevated urea, sacral skin discoloration or hairy nevus, tumor surface ulceration, inguinal lymphadenopathy, pulmonary metastasis, and absent anal opening [3,8]. In Sokoto, North-West Nigeria, Legbo et al reported the case of a newborn presenting with a fully formed third lower limb in the sacrococcygeal region [6]. The patient in our report presented with an infected gluteal mass associated with intermittent pain, bisphincteric incontinence, right lower limb wasting, and weakness.
SCTs may be associated with other congenital anomalies such as polydactyly, talipes calcaneovarus, encephalocele, anorectal malformation, omphalocele, sacral agenesis, syndactyly, and renal and cardiac malformations [4,9,17,18]. The differential diagnoses of SCTs are sacral myelomeningocele or meningocele, fibro-lipoma, embryonal rhabdomyosarcoma, anterior meningocele, neurofibroma, cystic hemangioma or lymphangioma, rectal duplication cysts, anal gland cysts, intracanalicular epidermoid tumors, dermal sinus stalks, hydromyelia, external Wilms tumors, retrorectal hamartomas, neuroblastomas, and pacinomas [4,8,9,17,19].
Clinically, Altman et al. classified SCTs as follows [20]:
- Type I: predominantly external with minimal presacral
component
- Type II: present externally but with significant intrapelvic extension
- Type III: external but predominant parts are intrapelvic
with abdominal extension
- Type IV: presacral with no external presentation.
Type I tumors are the most common worldwide and in Nigeria [3,4,10,11], just as in our patient.
Clinical assessment and radiologic imaging are the mainstay of diagnosis of SCTs [1,3,4,10,20]. Current investigation modalities include the use of Computed Tomography (CT) scans and MRI [8,17,21]. While CT scans delineate the lumbosacral and pelvic bony architecture well, MRI is better at delineating the soft tissue architecture. Pelvic and lumbosacral radiography, barium enema, and abdominopelvic ultrasonography are also routinely used, especially where the more current imaging modalities are unavailable or unaffordable [4,7,8,11,14]. Our patient’s presentation and treatment were delayed because of financial constraints; a philanthropic organization covered the costs of her investigations.
SCTs are diagnosed prenatally using routine 2-dimensional, advanced 3-dimensional and doppler ultrasonography, as well as ultrafast MRI [6,15,17,21], thus enabling anticipated delivery and early commencement of appropriate treatment. Treatment options include intra-uterine decompression of cystic teratomas [22] and planned early operative delivery of the fetus [17,21]. Early diagnosis and treatment of SCTs are important to prevent malignant transformation, estimated at 5% before one month of age, 60% by one year, and 75% after one year [5,7]. Our patient presented at 12 years of age without malignant tumor transformation.
Complete surgical excision is the treatment of choice for SCTs [3,4,6-9,15], and this can be done with excision of the coccyx to remove all nidi of pluripotent cells that could increase the risk of recurrence [10]. The approach could be through transabdominal, trans-perineal, trans-sacral, or combined abdomino-sacral routes [3,4,8,15]. Adjuvant or neoadjuvant radiotherapy or chemotherapy are known adjuncts to surgical excision, especially in malignant tumors [3-5,15,23]. Our patient had gross total en-bloc excision of the mass and overlying skin via a trans-sacral approach. The underlying defect was covered with mobilized muscle and skin flaps from the gluteal region. We did not consider adjuvant chemotherapy or radiotherapy because the histology was benign. Our approach is similar to procedures reported from other parts of Nigeria [3].
The outcome of the treatment of SCTs is good for benign tumors that are diagnosed and resected early [3,4,9]. Our patient’s histological diagnosis of type 1 immature teratoma is benign, and she has been symptom-free, with no recurrence, in four years of follow-up. In the series from Lagos, all 19 patients with benign tumors were alive at 2 years or more of follow-up, while all 11 patients with malignant tumors had tumor recurrence within 3 months and died within 12 months [3].
The postoperative complications include wound infection, wound dehiscence, post-operative diarrhea, post-operative hemorrhage, intraoperative rectal injury, and malignant recurrence [4,15], with a post-surgical mortality rate of 18% in neonates, and up to 55% in the post-neonatal period [9]. Our patient had pre-operative bowel preparation, with additional low rectal washout with soap and water enema on the morning of surgery as a precaution in case the tumor involved the rectum. This would prevent fecal contamination of the surgical wound and enable primary repair of any involved part of the rectum. However, she had transient urinary and fecal incontinence which resolved over time, and wound dehiscence which was managed with topical honey-impregnated dressings. As a consequence, her discharge from the hospital was delayed until 4 weeks post-surgery.
Conclusion
Sacrococcygeal teratomas are rare tumors but are the most common types of teratomas in childhood. Early diagnosis and prompt complete surgical excision prevent malignant transformation in benign tumors. This paper has discussed the case of a 12-year old girl with SCT who was successfully managed with complete en-bloc surgical excision in a secondary health facility in Nigeria. The case report shows that gross-total surgical excision of this rare tumor has an excellent prognosis for benign tumors, and can be successfully performed in are source-limited setting. The paper is limited by the fact that it is a single-patient case report and does not represent the full clinicopathological and outcome profile of this tumor in the Nigerian population. This highlights the need for a multi-institutional tumor registry in Nigeria through which detailed epidemiological, clinicopathological, management, and outcome studies on rare tumors, like SCT, can be conducted.
Disclosure: The authors declare no conflict of interest.
References
- Akang EE, Odunfa AO, Aghadiuno PU. Childhood teratomas in Ibadan, Nigeria. Human Pathology. 1992; 23: 449-453.
- Akang EE, Odunfa AO, Aghadiuno PU. A review of teratomas in Ibadan. African Journal of Medicine and Medical Science. 1994; 23: 53-60.
- Elesha SO, Aina AO, Odunjo EO. Sacrococcygeal teratomas in Lagos, Nigeria: Relationship of age, sex, clinical type, and histopathology to prognosis in 30 cases. East African Medical Journal. 1989; 66: 685-692.
- Abubakar AM, Nggada HA, Chinda JY. Sacrococcygeal teratoma in northeastern Nigeria: 18-years’ experience. Pediatric Surgery International. 2005; 21: 645-648.
- Mabogunje OA. Malignant childhood teratomas in the Nigerian savannah. Annals of Tropical Paediatrics, 1985; 5: 201-205.
- Legbo JN, Opara WE, Legbo JF. Mature sacrococcygeal teratoma: a case report. African Health Sciences, 2008; 8: 54-57.
- Onuoha CEO, Amah C, Ezike H. Managing sacrococcygeal teratoma in a newborn of a psychopathic widow: Case report. Nigerian Medical Journal. 2009; 50: 74.
- Afuwape OO, Ogundoyin OO, Ogunlana DI, Adeleye AO. Adult sacrococcygeal teratoma: A case report. Ghana Medical Journal. 2009; 43: 40-42.
- Nwako F, Ezi-Ashi I. Sacrococcygeal teratoma in Nigerian infants and children. International Surgery, 1974; 59: 402-405.
- Arigbabu SO, Ojikutu NA. Giant sacrococcygeal teratoma in an African child. Childs Nervous System. 1986; 2: 280.
- Nmadu PT. Sacrococcygeal teratoma in Zaria, Nigeria: Report of 47 cases. Annals of Tropical Paediatrics, 1995; 15: 299-302.
- Mabogunje OA, Nirodi NS, Lawrie JH, Edington GM. Teratomas in Nigerian children. East African Medical Journal. 1980; 57: 461- 469.
- Mabogunje OA, Nirodi NS, Harrison KA, Edington GM. Teratomas in adult Nigerians. African Journal of Medicine and Medical Sciences. 1980; 9: 151-158.
- Ozoilo K, Yilkudi M, Ede J. Sacrococcygeal teratoma in an adult female Nigerian. Annals of African Medicine. 2008; 7: 149.
- Chirdan L, Uba A, Pam S, Edino S, Mandong B, et al. Sacrococcygeal teratoma: Clinical characteristics and long-term outcome in Nigerian children. Annals of African Medicine. 2009; 8.
- Babatunde TO, Akang EEU, Ogun GO, Brown BJ. Pattern of childhood cancer in University College Hospital, Ibadan during 1991- 2010 and comparison with the previous three decades. Paediatrics and International Child Health. 2015; 35: 144-150.
- Alalfy M, Elebrashy A, Azmy O, Elgazzar A, Zakaria A, Gaafar H, et al. Review article: prenatal diagnosis and management of sacrococcygeal teratoma, a review of literature. ObstetGynecol Int J. 2019; 10: 47‒49.
- Anyanwu LJ, Mohammad A, Muhammad H, Aliyu I, Abdullahi L, Farinyaro A, et al. Schinzel-Giedon syndrome: A case with sacrococcygeal teratoma and cor-triatriatum dexter. Pan African Medical Journal. 2017; 26: 30.
- Nasir AA, Abdur Rahman LO, Ibrahim KOO, Adegoke MA, Afolabi JK, Adeniran JO. Giant plexiform neurofibroma mimicking sacrococcygeal teratoma. Journal of Surgical Technique & Case Report. 2012; 4: 50-52.
- Altman RP, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics surgical section survey. Journal of Pediatric Surgery. 1973; 9: 389-398.
- Gucciardo L, Uyttebroek A, de Wever I, Renard M, Claus F, Devlieger R, et al. Prenatal assessment and management of sacrococcygeal teratoma. Prenat Diagn. 2011; 31: 678–688.
- Garcia AM, Morgan III WM, Bruner JP. In utero decompression of a cystic grade IV sacrococcygeal teratoma. Fetal Diagn Ther. 1998; 13: 305–308.
- Abdur-Rahman L, Baba S, Bamigbola K, Olaoye A, Oyinloye A, Nasir A, et al. Outcome of management of complicated extragonadal teratoma in a resource poor setting. African Journal of Paediatric Surgery. 2013; 10.