
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
A case report and review of the literature on Von Recklinghausen’s disease complicated with cervicofacial plexiform neurofibromas
Hind Sahli*; Jihane El Mandour; Hassan En-nouali; Jamal El Fenni; Issam En-nafaa
Radiology Department, Mohammed V Military Training Hospital, Mohammed V University, Rabat, Morocco
*Corresponding Author: Hind Sahli
Radiology Department, Mohammed V Military
Training Hospital, Mohammed V University, Rabat,
Morocco.
Email: hind91sahli@gmail.com
Received : Feb 24, 2022
Accepted : Mar 22, 2022
Published : Mar 29, 2022
Archived : www.jcimcr.org
Copyright : © Sahli H (2022).
Citation: Sahli H, Mandour JE, En-nouali H, Fenni JE, En-nafaa I. A case report and review of the literature on Von Recklinghausen’s disease complicated with cervicofacial plexiform neurofibromas. J Clin Images Med Case Rep. 2022; 3(3): 1763.
Introduction
Neurofibromatosis type I, also known as Von Recklinghausen disease, is an autosomal dominant genodermatosis caused by a mutation in the NF1 gene on chromosome 17q11.2. It is characterized by a wide range of clinical signs and symptoms comprising macules of café-au-lait and tumours of the nerve sheath [1]. Neurofibromas make up 90% of all instances and plexiform tumors are less common [2]. Plexiform neurofibromas are largely pathognomonic for NF1, appearing as a “bag of worms” with diffuse involvement along a nerve segment and its branches [3]. The eyelids, trunk, and limbs are the most common sites for plexiform neurofibromas. It’s unusual to find localization in the cervico-facial region [4]. We present the case of a 20-year-old female patient who had been followed for von Recklinghausen’s disease from childhood and complained of a growing cervicofacial lump.
Case report
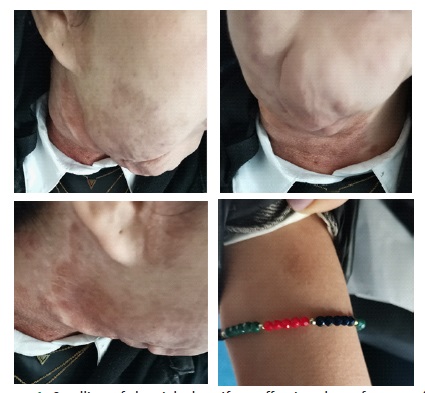
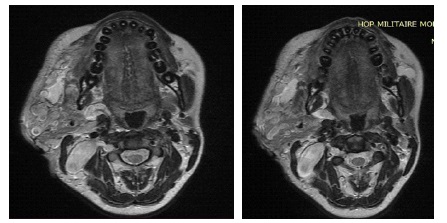
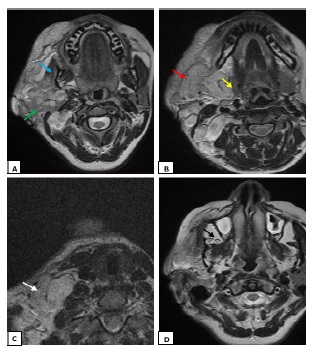
A 20-year-old female patient was seen for a swelling of the right hemiface measuring 8 cm in long axis. She was being followed for Von Recklinghausen neurofibromatosis. A tumor extending from the right parotid lodge to the homolateral supraclavicular fossa was discovered during the examination (Figure 1). On the right side, an MRI revealed a tortuous dilatation around the cervico-facial nerves and plexuses, with hypo signal T1 and hypersignal T2 enclosing a central region showing the cocarde aspect (Figure 2). These masses infiltrate on the right side the subcutaneous cellulo-fatty plane, the masseter muscle, the submaxillary and parotid glands, and fuse at the level of the infra temporal fossa without causing endocranial extension (Figure 3).
Discussion
Neurofibromatosis (NF) is a neurocutaneous syndrome characterized by skin, nervous system, bone, and endocrine gland abnormalities [5]. Plexiform neurofibromas are found in up to 30% of NF-1 cases, however they are less common in the craniomaxillofacial region [6]. These lesions usually appear early in childhood and progress to malignant peripheral nerve sheath tumors (MPNST) [6]. NF1 patients have a 16% five-year survival rate for MPNSTs, compared to 53% for non-NF1 patients [3].
Von Recklinghausen first characterized neurofibromatosis in two of his patients, Marie Kientz and Michael Bar, almost a century ago [1]. The NIH (National Institutes of Health Consensus Conference) identified four forms of neurofibromas: Cutaneous neurofibromas, subcutaneous neurofibromas, nodular neurofibromas, and diffuse plexiform neurofibroma [1]. The presence of at least two of the following criteria is required for a diagnosis of the disease: At least six café au lait spots that are larger than 5 mm before puberty and 15 mm after puberty, One or more plexiform neuromas, with two or more neurofibromas axillary and inguinal lentiginous spots; One optic tract glioma and two or more iris hamartomas (Lisch nodules); A bony lesion with a distinct appearance (pseudarthrosis of a long bone, spheno-orbital dysplasia, cervical kyphosis) [7].
Plexiform Neurofibroma (PFN) is a type of neurofibroma that can be nodular or diffuse [1]. The diffuse infiltration of nearby nerve trunks, cellulo fatty tissue, and muscle is explained by the non-encapsulated character [7]. It’s possible that it’ll erode the bone and invade the viscera [1].
Some authors recently identified four markers (epidermal growth factor receptor, interferon-y, interleukin-6, and tumor necrosis factor-) to differentiate between patients with NF-1 and healthy individuals, as well as two additional markers as potential early risk predictors of developing MPNST (InsulinLike Growth Factor Binding Protein 1 (IGFBP1) and Regulated Upon Activation, Normal T-Cell Expressed And Secreted) (RANTES) [6]. The scientists discovered that patients with NF-1 and MPNST had much higher amounts of these markers than those without [6].
Neurofibromas have different symptomatology depending on their topography. In the cervical region can compress the spinal cord or spread to the sympathetic or brachial plexus, resulting in Claude Bernard Horner syndrome or peripheral nerve paralysis [7].
Nerve sheath tumors can be diagnosed and characterized noninvasively using ultrasound, CT, and MRI [3]. Imaging is useful for a variety of reasons, including determining the extent of involvement and its impact on nearby structures, revealing related anomalies, and, last but not least, forecasting the possibility of malignant development [8].
On ultrasonography, most Peripheral Nerve Sheath Tumors (PNSTs) are hypoechoic with posterior acoustic enhancement, which can make them look like cystic lesions, abscess and a vascular malformation [3,8]. The target appearance may be seen with a hyperechoic center and hypoechoic periphery, corresponding to a fibrocollagenous zone in the center and a myxomatous region in the periphery [3,8]. Different forms of vascularization are revealed by the doppler color: moderate, central, or peripheral predominance [7]. Some neurofibromas may have poor blood flow.
The tomodensitometric analysis finds nodular, fusiform, or grappe lesions that are less dense than the muscle (20-30 UH) [7]. This low attenuation is due to the myelin-lipid composition, fat entrapment, and high water content in endoneurial myxoid tissue [8]. The importance of CT in assessing bone involvement and remodeling is crucial [8]
Due to its excellent contrast resolution and multiplanar capabilities, MRI is the most helpful imaging modality for characterizing tumor extent and suggesting neurogenic origin [3]. A homogeneously hyperintense T2 signal or the distinctive target sign with a central hypointense zone, oriented longitudinally in the nerve distribution, is diagnostic of peripheral nerve sheath tumours [7]. The enhancement can be central, diffuse, peripheral, or target-specific. Some MRI characteristics suggest potentially malignancy with 61% sensitivity and 90% specificity such as peripheral enhancement, a perilesional edema-like zone, intratumoural cystic lesions and the biggest dimension of the mass (greater than five centimetres) [3].
Imaging-based differential diagnoses include lymphatic or venous malformations, hemangiomas, and traumatic or inflammatory lesions [9].
Surgery is still the most common therapeutic option, the goal is to resect deforming masses and cancerous tissue when malignant transformation develops [6,9]. Good results have lately been described after the administration of interferon-gamma in unresectable, progressing, and symptomatic lesions [6].
References
- Plexiform neurofibroma encasing vital organs [Internet]. Indian Journal of Dermatology, Venereology and Leprology. 2009 [cité 17 janv 2022]. Disponible sur: https://ijdvl.com/plexiform-neurofibroma-encasing-vital-organs/
- Pérez Villena A, Duat Rodríguez A, García Peñas JJ, López Pino MA. Plexiform neurofibroma in an 8 year-old patient. Neurologia. 2013; 28: 319-320.
- Gosein M, Ameeral A, Banfield R, Mosodeen M. Plexiform Neurofibroma of the Wrist: Imaging Features and When to Suspect Malignancy. Case Reports in Radiology. 2013; 2013: e493752.
- Mahfoudhi M, Khaled K. Maladie de Von Recklinghausen compliquée de Neurofibromes plexiformes cervico-faciaux. Pan Afr Med J. 2015; 20: 408.
- Traoré B, Fofana Y. Le neurofibrome plexiforme diffus de la cuisse gauche chez une patiente âgée de 78 ans en milieu dermatologique à Bamako. Pan Afr Med J. 2017; 26: 82.
- Tchernev G, Chokoeva AA, Patterson JW, Bakardzhiev I, Wollina U, et al. Plexiform Neurofibroma. Medicine (Baltimore). 2016; 95: e2663.
- Bouimetarhan L, Bellamlih H, En-nafaa I, Fenni JE, Amil T, et al. Neurofibrome plexiforme cervical: à propos d’un cas. The Pan AfricanMedical Journal [Internet]. 2018; 30. Disponible sur: https://www.panafrican-med-journal.com/content/article/30/41/full
- Grover DrSB, Kundra DrR, Grover DrH, Gupta DrV, Gupta DrR, et al. Imaging diagnosis of plexiform neurofibroma- unravelling the confounding features: A report of two cases. Radiology Case Reports. 2021; 16: 2824-2833.
- Cacchione A, Carboni A, Lodi M, Vito RD, Carai A, Marrazzo A, et al. Congenital Craniofacial Plexiform Neurofibroma in Neurofibromatosis Type 1. Diagnostics (Basel). 2021; 11: 218.