
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Molecular analysis of SRD5A2 a novel homozygous variant found in a Mexican family
Morales Rios LC1; Calvo Anguiano G1; Martínez de Villarreal LE1; Villarreal Pérez JZ2; Castillo Castro C2; Lugo Trampe JJ1; García Castañeda GB1; López Uriarte GA1*
1 Department of Genetics, University Hospital “Dr. José E. González” UANL, Monterrey, Nuevo Leon 64460, Mexico.
2 Department of Endocrinology, University Hospital “Dr. José E. González” UANL, Monterrey, Nuevo Leon 64460, Mexico.
*Corresponding Author: López-Uriarte GA
Department of Genetics, University Hospital “Dr.
José E. González” UANL, Monterrey, Nuevo Leon
64460, Mexico.
Email: glopezu@uanl.edu.mx
Received : Mar 08, 2022
Accepted : Apr 08, 2022
Published : Apr 15, 2022
Archived : www.jcimcr.org
Copyright : © López-Uriarte GA (2022).
Abstract
5-alpha-Reductase deficiency is one of the causes involving disorders of sex development 46, XY. Mutations in SRD5A2 gene lead to a wide clinical manifestation, sometimes not giving a clear outlook for a clinical diagnosis, making molecular diagnosis an important diagnostic pillar of these conditions. Here, we show the importance of molecular and pedigree analysis for diagnosis and adequate genetic counseling.
Keywords: 46, XY disorder of sex development; ambiguous genitalia; 5-alpha-Reducatse type 2; novel mutation.
Abbreviations: T: Testosterone; DHT: Dihydrotestosterone; DSD: Disorder of Sex Development.
Citation: Rios MLC, Anguiano CG, Villarreal MLE, Pérez VJZ, López-Uriarte GA, et al. Molecular analysis of SRD5A2 a novel homozygous variant found in a Mexican family. J Clin Images Med Case Rep. 2022; 3(4): 1795.
Introduction
The steroid 5-alpha-Reductase type 2 is one of the isozymes that catalyze testosterone (T) to dihydrotestosterone (DHT) conversion, crucial androgen involved in normal external male genitalia differentiation [1]. The deficiency of this enzyme is related to an autosomal recessive disorder of sex development 46,XY (DSD XY), characterized by the presence of clitoral-like phallus, bifid scrotum, hypospadias, incomplete closure of the urogenital sinus, usually assigned as female [2].
SRD5A2 gene located on chromosome 2p23 contains 5 exons and encodes a 254 amino acid protein, is formed with 4 transmembrane regions and an androgen binding domain at the Nterminus. Around 100 mutations related to this condition have been described, being large deletions one of less common. The exons 1 and 4 are hot spots, and 60% of mutations appear in a homozygous state. All the mutations described to date affect enzymatic activity, producing the wide phenotype spectrum [3,4].
In Mexico has been reported a P212R substitution that is believed to be a founder variant in the Mexican population [5]. Nevertheless, in this study, we describe the molecular diagnosis of SRD5A2 gene in a Mexican family and analyze the family pedigree, finding a novel variant involving the exon 4.
Case presentation
A 24 years old female was referred to the Genetics Department for presenting primary amenorrhea and a 46,XY karyotype. She has a clinical history of bartolinitis at 12 years old, unilateral gonadectomy at 16 years old with a histopathological report compatible with testicular tissue. At physical exploration, we found no facial dysmorphism, short neck with acanthosis nigricans, breast development with a Tanner 2 stage, panniculus adipose in the abdomen, suprapubic surgical scar, fine pubic hair, labia majora covering the labia minora, clitoris with central urethral meatus, visible vaginal introitus and absence of axillary hair. The patient identifies as female and her sexual orientation is bisexual. Within laboratory and imaging studies total testosterone: 0.06 ng/mL (normal value: 0.084-0.481 ng/mL), DHT: 32 ng / dL (normal value: 3-24 ng/dL). Pelvic MRI: no evidence of uterus, vagina or gonads, suggestive data of prostate tissue and rudimentary seminal vesicles.
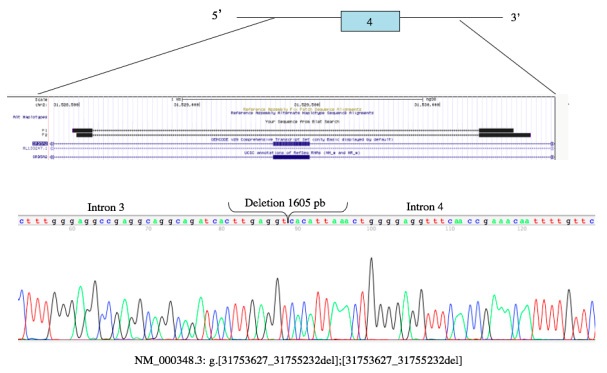
After performing the medical history and family pedigree a molecular analysis was made. It was performed to the whole family. Genomic DNA was extracted from peripheral blood lymphocytes with Puregene kit (© Qiagen) following the manufacturer’s instructions. Primer-BLAST (NCBI) was used to design primers. The coding regions of the SRD5A2 gene were amplified by PCR, and direct sequencing was performed using the BigDye Terminator v 3.1 Kit (Applied Biosystems) on an ABI3130 genetic analyzer (Applied Biosystems®). Primers were designed to amplify the ~ 50 bp flanking regions at both ends of each exon. Exon 4 did not amplify, primers flanking ~ 500, 1000 and 1500 bp were redesigned on both ends of exon 4 (introns 3 and 4). The generated sequences were compared with the reference sequence (NM_000348.4) (Figure 1).
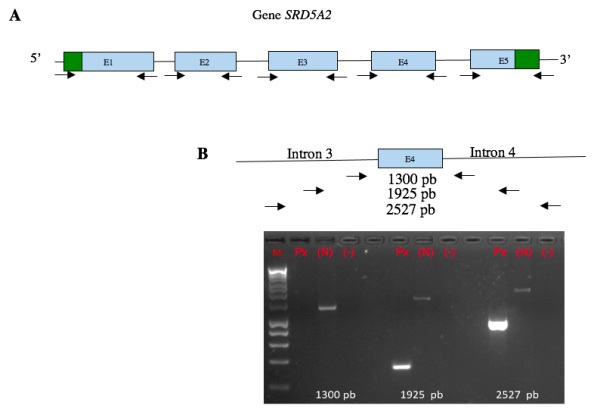
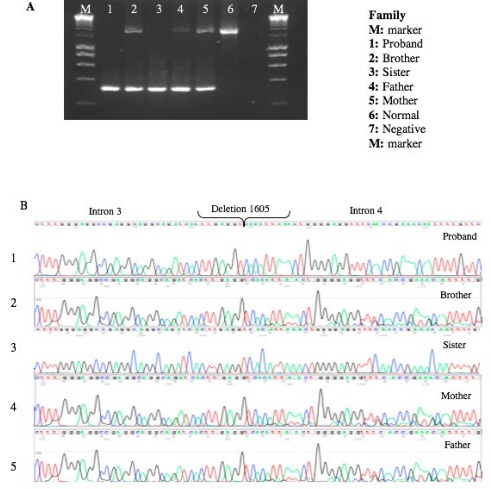
We found a 1605 bp deletion, which corresponds to amino acid 206 to 226, belonging to one of the transmembrane regions of the gene. Once we had localized the mutation, we search it directly in each family member (Figures 2,3).
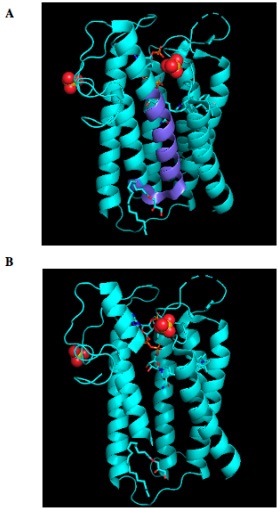
By using PyMOL, a molecular visualization system, we introduced the variant to analyze the impact in the conformation of the protein, we detect an almost whole helix loss (Figure 4).
Discussion
In our study, we analyze a family that comes from a small town in central Mexico that has a history of endogamy. Firstly, we began the approach with our proband, her clinical history was compatible with a DSD XY, the history of endogamy led us to begin with the molecular analysis of SRD5A2, being conscious of on differential diagnosis was androgen insensitivity.
During molecular analysis, we didn’t obtain an amplification of exon 4, the coverage towards introns was expanded to locate the cut-off point, identifying a deletion in a homozygous state, which has not been reported in the literature, nor in different databases (ClinVar, VarSome, Ensemble, HGMD) for what is considered a novel variant.
Once we had the molecular diagnosis, knowing that this condition is inherited as an autosomal recessive mode, we expanded the molecular analysis to the family, trying to find asymptomatic carriers (50%), affected individuals (25%), individuals without the mutation (25%). Doing the pedigree analysis, as expected, both parents were heterozygous carriers, like the brother. The sister of the patients with proven fertility has like our patient homozygous deletion, it has been described that females 46,XX are fertile, have a normal internal and external genitalia development, sometimes delayed menarche, reduce body hair [2].
The approach of patients with this condition doesn’t end with the molecular diagnosis, it is necessary to continue with complementary functional studies to determine the conformation of the protein and assess its function.
Conclusion
The implementation of a complementary molecular strategy for the analysis of the gene involved in DSD XY, which already had a clinical, biochemical and histopathological diagnosis, is of utmost importance to confirm the specific diagnosis, explain the pathophysiology, provide adequate transdisciplinary management of the patient and grant genetic counseling to her and her family.
Declarations
Conflict of interest: None declared.
Funding: Genetics Department University Hospital “Dr. José E. González,” UANL, Monterrey, Nuevo León, México.
References
- Thigpen AE, Davis DL, Milatovich A, Mendonca BB, ImperatoMcGinley J, et al. Molecular genetics of steroid 5 alpha-reductase 2 deficiency. J Clin Invest. 1992; 90: 799– 809.
- Kang HJ, Imperato-McGinley J, Zhu YS, Rosenwaks Z. The effect of 5α-reductase-2 deficiency on human fertility. Fertil Steril. 2014; 101: 310–316.
- Avendaño A, Paradisi I, Cammarata-Scalisi F, Callea M. 5-α-Reductase type 2 deficiency: is there a genotype-phenotype correlation? A review. Hormones. 2018; 17: 197–204.
- Nie M, Zhou Q, Mao J, Lu S, Wu X. Five novel mutations of SRD5A2 found in eight Chinese patients with 46,XY disorders of sex development. Mol Hum Reprod. 2011; 17: 57– 62.
- Vilchis F, Ramos L, Méndez JP, Benavides S, Canto P, Chávez B, et al. Molecular analysis of the SRD5A2 in 46,XY subjects with incomplete virilization: the P212R substitution of the steroid 5alpha-reductase 2 may constitute an ancestral founder mutation in Mexican patients. J Androl. 2010; 31: 358–364.