
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Asymptomatic patient: Incidental finding of lipoprotein glomerulopathy with ApoE Kyoto mutation
Cheuk-nam Ling*; Chun-hai Lo; Po-man Tsang; Shui-ying Cheng
Department of Pathology, United Christian Hospital, Hong Kong.
*Corresponding Author: Cheuk-nam Ling
Department of Pathology, United Christian Hospital,
Hong Kong.
Email: lcn467@ha.org.hk
Received : Mar 21, 2022
Accepted : Apr 15, 2022
Published : Apr 22, 2022
Archived : www.jcimcr.org
Copyright : © Cheuk-nam L (2022).
Abstract
Lipoprotein glomerulopathy is an unusual kidney disease with clinical presentation of proteinuria and renal insuffciency. The diagnosis is based on histological examination, featuring lipoprotein thrombi in glomerular capillaries. We present here an unusual case of lipoprotein glomerulopathy detected during pre-employment checkup. Histopathology examination confirmed the diagnosis, and subsequent genetic test revealed ApoE Kyoto mutation.
Keywords: Lipoprotein glomerulopathy; Kidney; Proteinuria.
Citation: Cheuk-nam L, Chun-hai L, Po-man T, Shui-ying C. Asymptomatic patient: Incidental finding of lipoprotein glomerulopathy with ApoE Kyoto mutation. J Clin Images Med Case Rep. 2022; 3(4): 1803.
Introduction
Lipoprotein glomerulopathy affects mostly individuals from Chinese and Japanese descent. Patients usually present at adulthood, but some may present at an earlier age. Around one third of the cases show slowly progressive disease. The clinical presentation includes proteinuria and renal insufficiency. Biochemical investigations typically reveal elevated serum apoE level. However, the diagnosis mainly relies on histological examination of the kidney, featuring lipoprotein thrombi in the capillary loops of glomeruli, while genetic testing shows mutations in apoE gene.
ApoE gene encodes apolipoprotein E. The latter are fat-binding proteins which bind to low-density lipoprotein receptors (LDLRs) for the catabolism of triglyceride-rich lipoproteins and their remnants. They are mainly produced by the liver and macrophages. Mutation of the apoE gene affects fat metabolism.
Lipoprotein glomerulopathy shows autosomal dominant inheritance with incomplete penetrance. Various mutations of apoE gene exist, of which most of them are missense variants adjacent to the low-density lipoprotein (LDL) receptor binding site. The histological features, despite the existence of multiple mutation variants, are similar. As described above, the presence of dilated glomerular capillary loops occluded by lipid thrombi, which are palely stained by PAS stain and are positively stained by Oil red O and Sudan stains, is a constant finding. Segmental double contouring of the glomerular basement membrane may also be identified. Electron microscopy shows that the thrombi are concentric lamellated electron-dense or lucent deposits with small lipid vacuoles. Deposition of the lipid material at the glomerular basement membrane may also be seen.
Pre-employment check-up is a common practice in Hong Kong. Most new graduates hired by commercial companies are provided with health check, including complete blood count, chest x-ray and urine dipstick. Here we present a case of lipoprotein glomerulopathy, discovered during pre-employment checkup.
Case report
A 24-year-old adult Chinese male was admitted into a local community hospital for renal biopsy. He was referred from a general practitioner who performed pre-employment check-up for the patient. The patient had no family history of renal disease. There was no significant past surgical or medical history. After admission, a series of blood tests were performed. The most significant abnormalities lied in the renal function test and lipid profile.
Other serological blood tests were performed, including screening for hepatitis B and C and autoantibodies (anti-nuclear antibodies, anti-dsDNA antibodies, anti-neutrophil cytoplasmic antibodies, and anti-glomerular basement membrane antibodies). The above tests were all negative. 24-hour urine protein showed 11.2 g/Day. After discussion with the patient, renal biopsy was performed for further investigation of the cause of his condition.
Gross examination
Three cores of tan biopsy tissue were submitted, each measured 1 cm in length. The tissue was sent for paraffin, frozen and resin block processing.
Microscopic examination
Paraffin section showed a total of thirty-one glomeruli, in which none was globally sclerosed. No segmental sclerosis was noted. All glomerular capillary lumina were occluded and distended by thombi-like material which were slightly vacuolated and lamellated in appearance. The thrombi were confined within the glomeruli and were weakly PAS positive and focally stained pale blue by Masson Trichrome (Figures 1 to 3). The glomeruli showed mild to moderate increase in mesangial matrix and cellularity. Silver stain showed no spikes or holes along the glomerular basement membrane. Focal segmental double contouring was noted. There were mild interstitial fibrosis and tubular atrophy. The renal interstitium showed no significant inflammation. The tubular epithelium, arterioles and interlobar arteries were unremarkable.
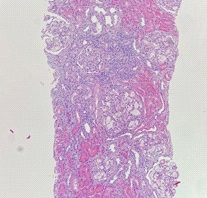
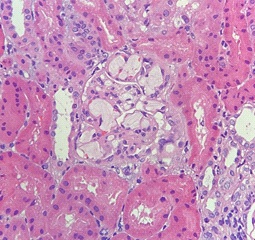
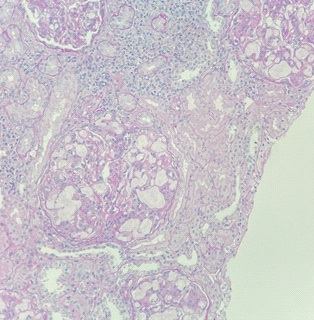
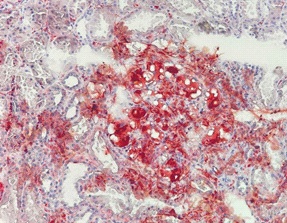
Oil red O was performed in the frozen section. The thrombi were stained positive (Figure 4). Direct immunofluorescence study showed trace mesangial staining for IgM. Staining for IgA, IgG, C1q, C3, fibrinogen were negative. No light chain restriction was demonstrated. No tubular basement membrane staining was noted. The overall features were consistent with lipoprotein glomerulopathy.
Electron microscopy
Electron microscopy was performed. Many of the glomerular capillaries were occluded by granular and amorphous thrombi and lipid vacuoles. Small to moderate amount of mesangial, subendothelial and intramembranous electron dense deposits were present. Focal segmental cellular interpositioning was seen. Some endothelial cells also showed lipid vacuoles. The glomerular basement membrane was thickened (400 to 520 nm) (Figures 5 and 6).

Molecular testing
The gene apoE (Apolipoprotein E) was tested. EDTA blood was used. The apoE gene were sequenced by the Sanger method. Heterozygous apoE NM_000041.4:c.127C>T, p.(Arg43Cys) (legacy name: p.(Arg25Cys)) was detected.
The gene apoE (Apolipoprotein E) was tested. EDTA blood was used. The apoE gene were sequenced by the Sanger method. Heterozygous apoE NM_000041.4:c.127C>T, p.(Arg43Cys) (legacy name: p.(Arg25Cys)) was detected.
Progress
Continuous clinical follow-up will be arranged in the future to monitor his progress. One year after the diagnosis, his kidney function gradually deteriorated. His father also received the genetic testing, and revealed the same mutation (ApoE Kyoto). However, his father had no proteinuria or renal derangement during subsequent examination and have remained asymptomatic.
Table 1: Laboratory investigations in year 2020 (the time of diagnosis) and year 2021 (current).
|
Year 2021 |
Year 2020 |
Creatinine |
109 |
165 (umol/L) |
eGFR |
57 |
69 (unit) |
Cholesterol |
10.2 |
6.7 (mmol/ L) |
Triglycerides |
2.8 |
1.4 (mmol/ L) |
HDL |
1.2 |
1.1 (mmol/ L) |
LDL |
4.8 |
7.8 (mmol/ L) |
Spot urine protein |
5.62 |
8.46 (g/L) |
Discussion
Here we have presented a case of young, Chinese man with lipoprotein glomerulopathy. Initially, the patient was asymptomatic and was not aware of the proteinuria until pre-employment health check-up.
The mutation discovered in our case was apoE Kyoto. It was originally described in a Japanese man with lipoprotein glomerulopathy. Most confirmed cases with such mutation are Asian, including Chinese and Japanese. In our case, a heterozygous missense mutation (C>T) in the apoE gene leading to an amino acid substitution of cysteine for arginine at codon 43 was detected. The apoEc.127C>T, p.(Arg43Cys) variant, also known as apo E Kyoto, has been reported in patients with biopsy-proven lipoprotein glomerulopathy and is involved in the etiology and pathogenesis of the disease. In vitro studies showed that this mutation is associated with a reduced binding capacity of apolipoprotein E to the LDL receptor, a decreased uptake by endothelial cells and subsequent glomerular lipoprotein deposition. Lipoprotein glomerulopathy appears to be an autosomal dominant condition caused by apoE mutation with incomplete penetrance. In our case, the same mutation was also detected in his father. Although the rest of his family, including his father, showed no clinical feature of proteinuria. Whole family targeted genetic testing is advised.
The apoE Kyoto mutation can lead to deposition of lipoprotein among glomerular vessels. The mutation reduces the capacity of apolipoprotein E to bind to LDL receptor. The uptake by endothelial cells is also decreased. Therefore, the anatomical pathologists should be aware of such changes. Apart from the alarming deposition of lipoprotein, one should also look for other possible associated changes, such as membranoproliferative glomerulonephritis (MPGN). The patients with lipoprotein glomerulopathy can be complicated by secondary MPGN, as there is expansile forces by the accumulation of lipoprotein. Co-existing immune complex glomerulopathy is also possible. Therefore, immunofluorescence is needed to rule out the coexisting entities. One case report recorded a case of lipoprotein glomerulopathy together with haemolytic uraemic syndrome, and they found a co-existing homozygous aHUS risk allele in that case [1].
Evidence has suggested that fibrates, a kind of lipid-lowering agent, help improve proteinuria and result in clinical remission. The suggested mechanism is that fibrates decrease the serum level of triglyceride-rich lipoproteins and their remnants which are composed of abnormal apoE [2]. On the other hand, corticosteroids, which are traditional treatment agents for nephrotic syndrome, are ineffective. For those patients who progress to end stage renal failure, renal transplantation is not a practical option as recurrence is inevitable.
Conclusion
Evidence has suggested that fibrates, a kind of lipid-lowering agent, help improve proteinuria and result in clinical remission. The suggested mechanism is that fibrates decrease the serum level of triglyceride-rich lipoproteins and their remnants which are composed of abnormal apoE [2]. On the other hand, corticosteroids, which are traditional treatment agents for nephrotic syndrome, are ineffective. For those patients who progress to end stage renal failure, renal transplantation is not a practical option as recurrence is inevitable.
References
- Kollbrunner L, Hirt-Minkowski P, Sanz J, Bresin E, Neuhaus TJ, Hopfer H, et al. Case Report: Lipoprotein Glomerulopathy Complicated by Atypical Hemolytic Uremic Syndrome. Front Med (Lausanne). 2021; 8: 679048. doi: 10.3389/fmed.2021.679048. PMID: 34150810; PMCID: PMC8206272.
- Zhangxue Hu, Songmin Huang, Yu Wu, Yunqiang Liu, Xiaoxia Liu, Dan Su, et al. Hereditary features, treatment, and prognosis of the lipoprotein glomerulopathy in patients with the APOE Kyoto mutation. Kidney Int. 2014; 85(2): 416-24. doi: 10.1038/ ki.2013.335. PMID:24025644. Epub 2013 Sep 11.
- Eduardo Cambruzzi, Karla Lais Pêgas. Pathogenesis, histopathologic findings and treatment modalities of lipoprotein glomerulopathy: A review. J Bras Nefrol. 2019; 41(3): 393-399. doi: 10.1590/2175-8239-jbn-2018-0148. PMID: 30421781. PMCID: PMC6788845.