
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Anesthetic management of syndactyly operation for a patient with apert syndrome: A case report
Begüm Nemika Gökdemir1*; Nedim Çekmen2
1 Researcher, Medical Doctor, Department of Anesthesiology, Faculty of Medicine, Baskent University, Fevzi Cakmak Caddesi, Ankara, Turkey
2 Professor, Department of Anesthesiology, Faculty of Medicine, Baskent University, Fevzi Cakmak Caddesi, Ankara, Turkey
*Corresponding Author: Begüm Nemika Gökdemir
Department of Anesthesiology, Faculty of Medicine,
Baskent University, Fevzi Cakmak Caddesi 10, Sokak
No:45 Bahcelievler, 06490 Ankara, Turkey.
Email: begokdemir@gmail.com
Received : Apr 06, 2022
Accepted : May 02, 2022
Published : May 09, 2022
Archived : www.jcimcr.org
Copyright : © Gokdemir BN (2022).
Abstract
Apert syndrome is autosomal dominant disease characterized by multiple craniofacial and limb deformities like syndactyly of feet and hands. There are many factors that challenging for anesthesiologists. In our case, a patient who has Apert syndrome came for syndactyly operation. We aim to highlight anesthetic considerations for Apert syndrome and how to maintain anesthesia during the operation.
Keywords: Apert syndrome; Syndactyly; Anesthetic management; Multidisciplinary approach.
Citation: Gökdemir BN, Çekmen N. Anesthetic management of syndactyly operation for a patient with apert syndrome: A case report. J Clin Images Med Case Rep. 2022; 3(5): 1825.
Introduction
Apert syndrome (AS) is a rare autosomal dominant congenital disorder characterized by craniosynostosis, high forehead, broad nose, maxillary hypoplasia, micrognathia, synostosis of cervical vertebrae, organ malformations and mental retardation and symmetric syndactyly of hands and feet. It was first reported by Wheaton in 1894 and a French pediatrician, Eugene Apert, published a series of nine cases in 1906 [1]. AS is estimated to affect 1 in 160,000 live births. It is most frequently caused by a de novo mutation. Two missense mutations in the fibroblast growth factor receptor 2 (FGFR-2) gene on chromosome 10 [2]. There are challenging factors for anesthesiologist such as preserving difficult airway, accessing difficult intravenous (IV) line, dosage calculations, airway hyperreactivity, temperature dysregulation. Due to craniofacial and skeletal anomalies, patients usually come for correction of extremity anomalies. In this case report, we present a 10-year-old girl with AS who came for second operation of syndactyly of hands.
Case report
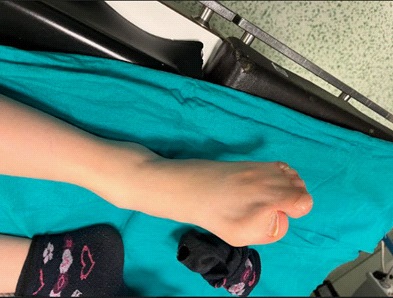
A 10-year-old girl came into operation room for syndactyly repairment. She has no comorbidities other than Apert syndrome. Her weight is 18 kg, American Society of Anesthesiologist (ASA) score was II. Her laboratory results, echocardiography and electrocardiogram were normal, but physical examination showed enlargement of the head and hypertelorism, flat and broad forehead, depressed nasal root, small mouth, micrognathia, and syndactyly of all extremities (Figures 1,2,3). Her mallampati grade was I, but because of her craniofacial anomalies, we predicted chance of difficult intubation and difficult airway cart which has different size of LMA (Laryngeal mask airway), endotracheal tubes, gum bougie, stile and videolaryngoscope was kept ready. General anesthesia was planned for the procedure.
Oral and written consent was obtained from the patient’s family before the operation. She was taken the operating room where standard monitoring was instated including non-invasive blood pressure, electrocardiogram, peripheral oxygen saturation, heart rate, end-tidal carbon dioxide and body temperature. We inserted 20 Gauge cannula for vein access. Additional IV line was placed after multiple attempts. Anesthesia induction was maintained with lidocaine 0.5 mg/kg, propofol 2 mg/kg, fentanyl 2 µcg/kg, rocuronium bromide 0.6 mg/kg. After 3 min, preoxygenation, with cricoid pressure was applied and we intubated the patient with 5.5 endotracheal tube using direct laryngoscope at first attempt. Due to craniofacial anomalies, difficult intubation cart kept ready. After intubation, we connected to mechanic ventilator with 4-6 mL/kg tidal volume and maximum 3-4 cm H2 O positive end expiratory pressure (PEEP). Anesthesia maintenance was provided by infusion of remifentanil 0.04-0.1 µcg/kg/min. and inhaler sevoflurane 2.2% in a 50% oxygen and air mixture. Fluid resuscitation was continued with 1/3 polydex due to normovolemia. Surgery was accomplished uneventful. Extubation was performed after she was able to breathe spontaneously. She was taken to the ward when she reached Aldrete score 8 from post anesthetic care unit (PACU).


Discussion
AS is one of the syndromes of craniosynostosis. It is characterized by the triad of craniosynostosis, midface hypoplasia and syndactyly of the hands and feet. These children require multiple surgical procedures for extremity anomalies and anesthesiologist faces multiple problems while dealing with them, such as airway management, securing IV access, calculating drug doses. If securing IV access is failed, an intraosseous or intramuscular route can be used. There is a risk of difficult mask ventilation and intubation due to mid face hypoplasia, cartilaginous abnormalities and angular deviation of the trachea, fusion of the cervical vertebra and tracheal stenosis [3]. If difficult intubation is suspected, a difficult intubation cart should be available in the operating room. Tracheotomy is compelling in children and can cause complications. Craniofacial anomalies are frequently associated with upper airway obstruction, especially during sleep, and can cause obstructive sleep apnea. Lack of development of tracheal cartilage may cause early death because of the obstruction [4]. Patient should be monitored postoperatively for risk of airway obstruction and nasopharyngeal airway must kept in the PACU.
These patients tend to be sweating and do not need to be warming when undergoing syndactyly surgery. Hyperpyrexia may develop if they are overwarmed, so temperature should always be monitored. If there is a risk for hypothermia due to loss of blood or coldness of the theatre room, warm air blankets and fluid warmers can be used.
These children might have profuse and dense secretions which can result in wheezing and increased airway irritability. Atropine has been suggested as a premedication to reduce secretions [5]. We must be vigilant in case of EtCO2 values increases in mechanic ventilator. It should be kept in mind that it may be a mucus plug like the case of Basar et al. [6]. Postoperative respiratory physiotherapy may reduce postoperative complications. Hudson, et al. reported that a-7-month-year-old male infant during bronchoscopy and tracheostomy due to airway malformations suddenly developed hypoxemia, therefore they used rigid bronchoscopy for correction of endotracheal tube. In our case, we considered airway complication odds so difficult airway cart was kept ready [7].
There is no consensus between general or regional anesthesia and there are no known contraindications to specific anesthetic agent and drug [5]. Regional anesthesia should be used whenever possible, since incidence of obstructive sleep apnea is higher in these patients. Moreover, regional anesthesia reduces perioperative opioid requirements and therefore the opioid side effects like deep sedation, respiratory depression and nausea and vomiting were less common than general anesthesia. Despite its positive aspects, regional anesthesia can be difficult for anatomical reasons owing to anatomical variations in shoulder joint and related structures and ultrasound guidance may help in these patients [8].
Conclusion
Anesthetic management of a patient with AS is compelling for anesthetist. So, comprehensive preoperative evaluation and careful anesthesia plan are imperative for these population. Because of the complexity of the syndrome a multidisciplinary (cerebral, respiratory, maxillofacial spinal and orthopedic) approach and managing accordingly are essential in treating the psychological, aesthetic and functional problems.
Declarations
Conflict of Interest: None.
Financial disclosures: None.
References
- Fadda MT, Ierardo G, Ladniak B, Di Giorgio G, Caporlingua A, Raponi I, et al. Treatment timing and multidisciplinary approach in Apert syndrome. Ann Stomatol. 2015; 6(2): 58-63.
- Ciurea A.V, Toader C. Genetics of craniosynostosis: review of the literature. J Med Life. 2009; 2(1):5-17.
- Barnett S, Moloney C, Bingham R. Perioperative complications in children with Apert Syndrome: A review of 509 anesthetics. Pediatr Anesth. 2011; 21:72-77.
- Papay FA, McCarthy VP, Eliachar I, Arnold J. Laryngotracheal anomalies in children with craniofacial syndromes. J Craniofac Surg. 2002; 13:351-364.
- Bansal, Teena & Jaiswal, Rajmala & Hooda, Sarla & Mangla, Pardeep. Apert syndrome: Anaesthetic concerns and challenges. Egyptian Journal of Anaesthesia. 2015; 31:85-87.
- Basar H, Buyukkocak U, Kaymak C et al. An intraoperative unexpected respiratory problem in a patient with Apert syndrome. Minerva Anestesiol. 2007; 73: 603-606.
- Hutson LR, Young E, Guarisco L. Tracheal anomalies complicating ventilation of an infant with Apert syndrome. Journal of Clinical Anesthesia. 2007; 19: 551-554.
- Chan VWS, Perlas A, McCartney CJL, Brull R, Xu D, Abbas B. Ultrasound guidance improves success rate of axillary brachial plexus block. Can J Anaesth. 2007; 54: 176-182.