
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Trisomy 18 and microdeletion 18p mosaicism: A case report and literature review
Wei Tang1 ; Xiao-Ying Wang1,2; Chao-Chun Zou1*
1 Department of Endocrinology, the Children’s Hospital of Zhejiang University School of Medicine, China.
2 Department of Pediatrics, the First people’s Hospital of Jiande, China.
*Corresponding Author: Chao-Chun ZOU
The Children’s Hospital of Zhejiang University
School of Medicine 3333 Binsheng Road, Hangzhou
310051, China.
Email: zcc14@zju.edu.cn
Received : Apr 25, 2022
Accepted : May 24, 2022
Published : May 31, 2022
Archived : www.jcimcr.org
Copyright : © Chao-Chun Z (2022).
Abstract
The trisomy 18 syndrome is a common chromosomal disorder due to the presence of an extra chromosome 18, either complete, mosaic trisomy or partial trisomy 18q. The mosaic trisomy 18 patients’ phenotype was extremely variable, from the absence of dysmorphic features to complete trisomy 18 syndrome. The phenotype of 18p deletion syndrome is variable and almost all survived. A 2-year-old girl was referred to our hospital due to growth delay. Mild dysmorphy including thin hair, frontal bossing, low set ears, broad-flat nose, nostrils slightly upward, downturned corners of the mouth, dysplasia teeth, small hands and fingers bilaterally was observed. The karyotype of peripheral leukocyte showed 46,XX, psu idic (18)(p11.2)[55]/46,XX, del (18)(p11.2) [45]. We report this case to add to our knowledge of the trisomy 18 and microdeletion 18p mosaicism.
Keywords: Trisomy 18; mosaic; 18p microdeletion; Psychomotor retardation; Karyotype.
Citation: Tang W, Xiao-Ying W, Chao-Chun Z. Trisomy 18 and microdeletion 18p mosaicism: A case report and literature review. J Clin Images Med Case Rep. 2022; 3(5): 1861.
Introduction
The trisomy 18 syndrome was first reported by Edwards et al in 1960, also known as Edwards syndrome [1]. It is the second most common autosomal chromosomal disorder after trisomy 21 (Down’s syndrome) due to the presence of an extra chromosome 18, which has three basic types: complete, mosaic and partial type [1-3]. The syndrome presents a recognizable pattern of major and minor anomalies, significant psychomotor and cognitive disability are associated with high neonatal and infant morbidity and mortality. The estimated overall prevalence of trisomy 18 in live born is approximately 1/6,000 to 1/8,000 while the incidence in fetus is much higher, the difference is caused by fetal loss and pregnancy termination after prenatal diagnosis [2,4]. The mosaic trisomy 18 usually means having more than one cell line in the individual, and it occurs in approximately 5 percent in all trisomy 18 patients [5]. The phenotypic manifestations are highly variable, from the absence of dysmorphic features to the complete trisomy 18 syndrome [6].
Since the clinical outcomes of complete and mosaic trisomy 18 can be different, it is of vital importance to achieve a correct diagnosis because of implications in medical management and genetic counselling.
18p deletion was first described by de Grouchy and colleagues in 1963 and was estimated to occur in approximately 1/50,000 live born, which results from deletion of a part or full of the short arm of chromosome 18 [7]. The mostly reported clinical features include cognitive impairment, congenital heart defects, small stature, minor facial dysmorphy, and skeletal deformities [7-10]. Typical facial features include hypertelorism, ptosis, strabismus, broad–flat nose, micrognathia, and low-set big ears. Holoprosencephaly may be seen in approximately 10–15% of patients [7]. In addition, speech and language difficulties, pituitary abnormalities, generalized seizures, dystonia, and autoimmune diseases have also been described [7,11-13]. However, these non-specific features are easily overlooked clinically. The clinical phenotype severity is related to the size and location of deletion region. In this report, we present a 2-yearold girl of mosaic trisomy 18 and 18p microdeletion with mild psychomotor retardation, cognitive impairment and language developmental disability.
Case description
A 2-year-old female second child of non-consanguineous parents was admitted to our hospital due to growth delay. Her mother and father were 34 years old and 38 years old when giving birth to her. She was born at full-term with uncomplicated gestation, her birth weight was 3.35 kg and the length was about 50 cm. No feeding difficulty and complications were referred in the neonatal period. She had a motor retardation of autonomous walking until 22-months old and intelligence disability and language disability. She only knew a few simple words like ”mama“, not ”baba“, and she cannot communicate clearly with others though she was willing to speak to strangers. Gesell Developmental Schedules performed in local hospital indicated mental developmental delay in motor behavior, language behavior, adaptive behavior and personal-social behavior at age of one year and 8 months old. The height of her father, mother and 15 years old sister were 165 cm, 161 cm, and 155 cm, respectively. No similar history was noted in her family.
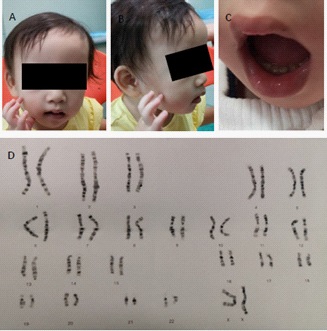
On physical examination, she had a height of 81.4 cm below -3SD and a weigh of 11.6 kg below -1SD. Mild craniofacial dysmorphy was present, including thin hair, frontal bossing, low set ears, broad-flat nose, nostrils slightly upward, and downturned corners of the mouth while other craniofacial anomalies were not obvious (Figure 1A). Her hands were small especially her fingers, but the fingernails are normal (Figure 1B). Her teeth were dysplasia (Figure 1C). The echocardiography revealed patent foramen ovale (ϕ 2.96 mm) while no murmur was present. The muscle tension was normal and no other organ abnormality was detected in our patient.
Laboratory examinations (urine, liver, kidney, thyroid hormone, GS/MS and blood glucose analyses) were all normal. Insulin-like growth factor-1 was 72.5 ng/ml (normal range, 55- 327 ng/ml).
Management and outcome
Ten months ago, the child was brought to a local hospital with developmental delay, the peripheral leukocyte karyotype was taken and revealed two abnormal cell lines, the result was 46,XX, psu idic (18)(p11.2)[55]/46, XX, del (18)(p11.2)[45]. She was then referred to another hospital to take the whole-exome sequencing demonstrating a deletion at 18p11.32-p11.22 (GRch37/hg19, chr18:158679 9708482del) and a duplication at 18p11.21-q23(GRch37/hg19, chr18:12012132 78005255dup). She was diagnosed mosaic trisomy 18 syndrome.
Discussion
The first reported patients with trisomy 18 syndrome were initially described by Edwards et al and Smith et al in 1960s [1,14], while the first case of mosaic trisomy 18 was reported in 1965 [15]. Less than 5% portion of patients have mosaicism of trisomy 18, and Banka et al reminded that routine karyotype from lymphocyte culture may not be sufficient to diagnose mosaicism if practitioners suspect a diagnosis of mosaic trisomy 18, karyotype from skin fibroblasts should be considered [16]. Since then over 40 cases of mosaic trisomy 18 have been described, Tucker et al reviewed 33 reported individuals of mosaic trisomy 18 and added 2 more cases in 2007 [6]. Their clinical manifestations are extremely variable from complete trisomy 18 syndrome with early death to near totally normal [2,6]. Some physical features are relatively more common and included brachydactyly, high arched palate, microcephaly, delayed bone age, frequent respiratory infections and otitis media, heart defect, 5th finger clinodactyly, micrognathia, and hypotonia. The most common heart defect is ventricular septal defect in mosaic trisomy 18. Our case has mild craniofacial dysmorphy and patent foramen ovale, and no other physical anomalies were observed.
Trisomy 18 mosaicism usually indicates the existence of more than one cell line in the individual. The peripheral leukocyte karyotype demonstrates pseudodicentric chromosome substituting a normal chromosome 18 in 55 cells and chromosome 18 missing the end of the short arm in 45 cells. The skin fibroblasts karyotype was not taken. Furthermore, there is no correlation between the physical and intellectual findings and the percentage of trisomy 18 cells in either peripheral leukocytes or skin fibroblasts [6,16]. Besides, there is no correlation between the percentage of trisomic cells in peripheral leukocytes and brain, gonads, or other key organs [6]. The variety of mosaic trisomy 18 may be related to the percentage of trisomic cells in different key organs of the body.
For complete trisomy 18 patients, approximately 50% of infants live longer than one week and about 5-10% of children survive beyond the first year [2]. In overall, trisomy 18 mosaicism patients usually survive longer when compared to complete trisomy 18 [6,17]. This does not mean that all the mosaic trisomy 18 patients have a longer survival, some died a few hours after birth. For normal or mild phenotypical mosaic trisomy 18 cases, some were diagnosed due to recurrent miscarriages or giving birth to a child with trisomy 18 while others may never be identified [6]. 18p deletion syndrome, also called monosomy 18p and De Grouchy syndrome type I, which means a deletion of full short arm of chromosome 18 or a microdeletion of the short arm of chromosome 18. Some researches showed that nearly half of patients have breakpoints in the centromeric region and the rest scatter in the short arm, and approximately half of the deletions occur on the maternal chromosome 18 no matter where the breakpoint locations are [9,18]. Our case’s breakpoint is at the 18p11.32-p11.22. Approximately two thirds of patients’18p deletion are de novo; the rest may be due to a de novo unbalanced translocation or malsegregation of parental chromosome rearrangement or a ring chromosome [19]. The patient’s height and weight is 81.4 cm below -3SD, 11.6 kg below -1SD, respectively. It may be a prodrome of small stature, but her insulin-like growth factor-1 was normal. It also could be contributed to feeding problem. More follow-up work needs to be done to figure it out. Some reported cases show that growth hormone replacement treatment is efficient in growth hormone deficiency patients [8].
Our case has trisomy 18 and microdeletion 18p mosaicism simultaneously. The possibility of meiotic chromosomal nondisjunction of the ovogonia/spermatocyte was increased because of her parents’ advanced maternal age, some women may have higher a risk for nondisjunction [20]. More possible mechanism may be a de novo unequal recombination occurring in early embryonic mitosis. Some deletions are from the parents [9], there is no way to figure her mutation mechanism out since we can not get her parents’ consent to analysis. The phenotype of our case combines two syndromes’ typical features, including common psychomotor retardation, cognitive impairment and congenital heart defect, characteristic small stature and language impairment of 18p deletion syndrome. Our case’s uncharacteristic craniofacial features also combine two syndromes.
Conclusion
In a conclusion, mosaic trisomy 18 and 18p deletion syndrome both are chromosomal disorders which has a variety of clinical manifestations. If an individual has untypical phenotypical anomalies and psychomotor and cognitive disability, chromosome disorder should be considered and cytogenic analysis is needed.
Declarations
Acknowledgement: We thank the patient and his parents for permitting us to use the data.
Funding: This research was supported by the key R & D Projects of Zhejiang Provincial Department of Science and Technology (2021C03094), National Natural Science Foundation (81371215 and 81670786).
Availability of data and materials: The datasets generated and/or analyzed during the current study are available.
Competing interests: There is no any competing interest.
Authors’ contributions: CCZ conceptualized and designed the study, and reviewed and revised the manuscript. WT drafted the initial manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Informed consent: We submitted an ethics application to the hospital, which included an application of excepting informed consent. Because the case took a long time, it was difficult to get in touch, and the identifiable information of the patient and family was not exposed. We have attached the PDF of the approval letter of ethics of our hospital, it should have the same legal effect of informed consent.
References
- Edwards JH, Harnden DG, Cameron AH, Crosse VM, Wolff OH. A new trisomic syndrome. Lancet. 1960; 1: 787-90.
- Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet J Rare Dis. 2012; 7: 81.
- Mudaliyar US, Mudaliyar SU. Strawberry skull in Edwards syndrome. BJR Case Rep. 2017; 3: 20170045.
- Rasmussen SA, Wong LY, Yang Q, May KM, Friedman JM. Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics. 2003; 111: 777-84.
- Fitas AL, Paiva M, Cordeiro AI, Nunes L, Cordeiro-Ferreira G. Mosaic trisomy 18 in a five-month-old infant. Case Rep Pediatr. 2013; 2013: 929861.
- Tucker ME, Garringer HJ, Weaver DD. Phenotypic spectrum of mosaic trisomy 18: two new patients, a literature review, and counseling issues. Am J Med Genet A. 2007; 143A: 505-17.
- Turleau C Monosomy 18p. Orphanet J Rare Dis. 2008; 3: 4.
- Xiao B, Ji X, Ye H, Liu Y, Cao Y, Sun Y, et al. Genotypic and phenotypic analysis of a patient with de novo partial monosomy 18p and partial trisomy 18q. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2019; 36: 484-487.
- HasI-Zogaj M, Sebold C, Heard P, Carter E, Soileau B, Hill A, et al. A review of 18p deletions. Am J Med Genet C Semin Med Genet. 2015; 169: 251-64.
- Yi Z, Yingjun X, Yongzhen C, Liangying Z, Meijiao S, Baojiang C. Prenatal diagnosis of pure partial monosomy 18p associated with holoprosencephaly and congenital heart defects. Gene. 2014; 533: 565-9.
- Rao VB, Kerketta L, Korgaonkar S, Ghosh K, Mohanty D. 18p deletion syndrome with a 45, XY, t (14; 18) (p11;q11.2), -18, karyotype. Ann Genet. 2001; 44: 187-90.
- Graziadio C, Rosa RF, Zen PR, Pinto LL, Barea LM, Paskulin GA. Dystonia, autoimmune disease and cerebral white matter abnormalities in a patient with 18p deletion. Arq Neuropsiquiatr. 2009; 67: 689-91.
- Mcgoey RR, Gedalia A, Marble M. Monosomy 18p and immunologic dysfunction: review of the literature and a new case report with thyroiditis, IgA deficiency, and systemic lupus erythematosus. Clin Dysmorphol. 2011; 20: 127-30.
- Smith DW, Patau K, Therman E, Inhorn SL. A new autosomal trisomy syndrome: multiple congenital anomalies caused by an extra chromosome. J Pediatr. 1960; 57: 338-45.
- Hook E B, Yunis JJ. Congenital asymmetry associated with trisomy 18 mosaicism. Am J Dis Child. 1965; 110: 551-5.
- Banka S, Metcalfe K, Clayton-Smith J. Trisomy 18 mosaicism: report of two cases. World J Pediatr. 2013; 9: 179-81.
- WU J, SPRINGETT A, MORRIS JK. Survival of trisomy 18 (Edwards syndrome) and trisomy 13 (Patau Syndrome) in England and Wales: 2004-2011. Am J Med Genet A. 2013; 161A: 2512-8.
- Schaub RL, Reveles XT, Baillargeon J, Leach RJ, Cody JD. 2002. Molecular characterization of 18p deletions: evidence for a breakpoint cluster. Genet Med. 2002; 4: 15-9.
- Koshy B, Mandal K, Srivastava VM, Loius PT, Danda S. Familial 18p deletion syndrome and 18p partial trisomy inherited from a mother with balanced translocation. Clin Dysmorphol. 2011; 20: 148-51.
- Warburton D, Dallaire L, Thangavelu M, Ross L, Levin B, Kline J. Trisomy recurrence: a reconsideration based on North American data. Am J Hum Genet. 2004; 75: 376-85.