
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Kikuchi Fujimoto disease: A rare case of recurrence
Júlia Henriques1; Ana Paula Azevedo1,2*; Filipa Mousinho3; Francisca Miranda3; Paula Sousa e Santos4; Martinha Chorão5; Ana Batalha Reis1; Cândido Silva1; Susana Matos1; Flora Meireles1; João Viana1
1Clinical Pathology Department, Hospital of São Francisco Xavier, West Lisbon Hospital Centre, 1449-005 Lisbon, Portugal.
2Centre for Toxicogenomics and Human Health (ToxOmics), Genetics, Oncology and Human Toxicology; NOVA Medical School / Faculty of Medical Sciences, NOVA University of Lisbon, 1169-056 Lisbon, Portugal.
3Clinical Hematology Department, Hospital of São Francisco Xavier, West Lisbon Hospital Centre, 1449-005 Lisbon, Portugal.
4Clinical Hematology Department, Hospital of Santo António dos Capuchos, Central Lisbon Hospital Centre, 1169-050 Lisbon, Portugal.
5Pathology Department, Hospital of Egas Moniz, West Lisbon Hospital Centre, 1449-005 Lisbon, Portugal.
*Corresponding Author : Ana Paula Azevedo
Clinical Pathology Department, Hospital of São
Francisco Xavier, West Lisbon Hospital Centre, Estrada do Forte do Alto do Duque, 1449-005 Lisbon,
Portugal.
Email: apazevedo@chlo.min-saude.pt
Received : May 21, 2022
Accepted : Jun 21, 2022
Published : Jun 28, 2022
Archived : www.jcimcr.org
Copyright : © Azevedo AP (2022).
Abstract
Background: Kikuchi-Fujimoto’s Disease (KFD) is rare, benign and more frequent in Asia. It is characterized by the appearance of lymphadenopathies and fever. Histological examination of the lymph nodes make the diagnosis. It is a self-limited disease. Relapses rarely occur.
Case presentation: We described a case in Europe, with the particularity of having presented recurrence.
Man, 66-year-old, leukodermic, admitted for weight loss, fever, multiple lymphadenopathies, hepato-splenomegaly, severe bicytopenia and acute kidney injury. Infectious, autoimmune and haematological diseases were excluded. Histological examination of cervical lymphadenopathy was compatible with necrotic KFD. After corticosteroid therapy, the condition resolved. Two years later, he was hospitalized for similar symptoms, hepato-splenomegaly, mediastinal and lumboaortic lymphadenopathies. Recurrence of KFD was admitted
Conclusions: This case report is intended to alert to the difficulty of diagnosing a rare cause of lymphadenopathies in the western world. The diagnosis isn´t obvious, required the exclusion of other, much more frequent causes and whose treatment is more specific.
Keywords: Kikuchi-Fijimoto; Necrotizing lymphadenitis; Fever; Recurrence.
Abbreviations: CMV: Cytomegalovirus; CT scan: Computed tomography scan; EBV: Epstein Barr Virus; HAV: Hepatitis A; HBV: Hepatitis B; HCV: Hepatitis C virus; HIV: Human Imunodeficiency Virus; HHV8: Human Herpes Virus 8; IGRA: Interferon Gamma Release Assay; KFD: Kikuchi Fujimoto’s Disease; NSAIDs: Nonsteroidal Antiinflammatory Drugs; SLE: Systemic Lupus Erythematosus.
Citation: Henriques J, Azevedo AP, Mousinho F, Miranda F, Santos PS, et al. Kikuchi Fujimoto disease: A rare case of recurrence. J Clin Images Med Case Rep. 2022; 3(6): 1913.
Background
Kikuchi-Fujimoto Disease (KFD) is the most common cause of histiocytic necrotizing lymphadenitis (non-granulomatous inflammation and necrosis of the lymph node), followed by systemic lupus erythematosus - SLE and acute Epstein Barr virus - EBV lymphadenitis [1,2]. It is a rare disease that was described for the first time in Japan, independently by Kikuchi and Fujimoto, in 1972 [1,2]. It is a benign and self-limiting condition characterized by regional lymphadenopathy (usually posterior cervical, in 60-90% of cases) associated with fever, and other signs and symptoms such as cutaneous lesions, arthritis, fatigue and hepatosplenomegaly [3,4]. In rare cases, neurologic complications including meningoencephalitis, cerebellar ataxia, encephalitis with central nervous system lesions, and most commonly aseptic meningitis have been described [5].
Etiology is still unknown, but it is thought to be related to previous viral infections or autoimmune processes [6].
While having a world-wide distribution, it is more common in Japan and in other asiatic regions [2], usually affecting young women in the third decade of life, with a ratio female to male of 4:1 [3,7]. However, rare cases have been described in pediatric, most commonly in males, and geriatric populations [8-10].
This disorder is of particular clinical importance, because it can mimic and may often be misdiagnosed (rate up to 40%) [11] with other more common or severe diseases, such as viral infections, tuberculosis, toxoplasmosis, sarcoidosis, SLE, reactive lymphadenitis, lymphoma and metastatic conditions [3,6,12]. Moreover, when associated with other conditions (mainly SLE) KFD can follow an even more severe evolution [13].
This overlapping similarity make it a diagnostic challenge, which is based on histology of the affected lymph nodes [2].
Most patients have a self-limited disease course, acute to subacute, with clear improvement after a few months, with a low recurrence rate of 3-4% [11,14,15].
Usually, treatment is not required, but for patients with severe symptoms, short-term administration of corticosteroids and Nonsteroidal Anti Inflammatory Drugs (NSAIDs) may be effective [16,17], and long-term follow-up is necessary, due to the possibility of recurrence [16].
Case presentation
A 66-year-old Caucasian Portuguese man was admitted to the emergency department with asthenia, anorexia, progressive weight loss (15 kg in 2 months) and fever. Past medical history included gout medicated with allopurinol, and marked smoking (about 50 cigarette/day) and alcoholic habits. No relevant family history was present.
On physical examination, he was dehydrated and with skin palor, presenting multiple bilateral lymphadenopathies on submandibular, posterior cervical and inguinal regions, and palpable hepatosplenomegaly. There was no evidence of limbs edema, but the patient had pain and cyanosis of the second finger of the left hand, suggesting an ischemic process. The remaining physical examination was normal.
The blood tests showed severe bicytopenia (hemoglobin-5.6 g/dL and platelets-7.0x109 /L), acute kidney injury (creatinine-7.7 mg/dL) with oligoanuria and need for dialysis, and hyperuricemia (23 mg/dL). The patient was hospitalized for aetiological investigation of lymphoproliferative disease vs infectious/autoimmune condition
Autoimmunity screening was unremarkable (rheumatoid factor, anti-neutrophil cytoplasmic, anti-DNAds, anti-nuclear, anti-Ro/La/Sm/RNP/Scl70/Jo1, anti-glomerular basement membrane and anti-phospholipid antibodies). Serologies for infectious agents (EBV, Cytomegalovirus (CMV), Rubella, Hepatitis B and C vírus (HBV and HCV, respectively), Human Imunodeficiency Virus (HIV), Human Herpes Virus 8 (HHV8), Treponema pallidum and Toxoplasma gondii), as well as the evaluation of tumor markers (total PSA, CEA, AFP, CA19.9, CA15.3, CA125) were negative. Additional laboratory tests showed sedimentation rate higher than 120 mm/h, adenosine deaminase 56.3 U/L (RV: 6.8 - 18.2), angiotensin converting enzyme 43.5 U/L (RV: 8 - 52), β2 microglobulin: 52,7 mg/L (RV: 1,09 - 2,53), and IL-6: 349,6 pg/mL (RV: < 17).
Immunofixation of serum proteins showed the presence of oligoclonal gammopathy compatible with an infectious/inflammatory condition.
Bone marrow aspirate revealed plasmocytosis (20%) with some dysmorphic signs, and very large cells with a high nuclear/cytoplasmic ratio, fine chromatin, basophilic cytoplasm with vacuoles suggesting a lymphoid origin. According to these findings, two diagnostic hypothesis were considered, namely a reactive condition vs a plasma cell dyscrasia. Immunophenotyping by flow cytometry did not detect abnormal lymphoid, nor plasma cell populations, norincreased number of blasts. Bone marrow biopsy evidenced rareatypical lymphoid foci suggestive of infiltration by lymphoproliferative disease, hyperplasia of hematopoietic lines and exuberant reactive pattern plasmacytosis. Karyotype analysis was in conclusive
Computed Tomography Scan (CT-scan) of the neck, chest and abdomen revealed mediastinal and bilateral lumbar-aortic lymphadenopathies and mild to moderate hepatosplenomegaly
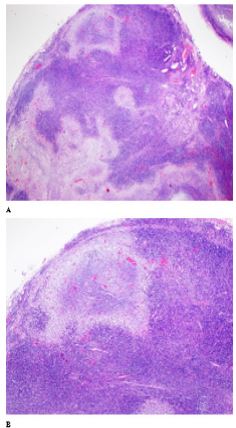
In an attempt to better clarify the observations made in the bone marrow biopsy, an excisional biopsy of right cervical lymphadenopathy was performedand the histological examination was compatible with necrotizing lymphadenopathy with severe plasmacytosis (Figure 1). Immunophenotyping by flow cytometry was normal.
One month after hospitalization, fever was persistent, although cultural tests (urine, blood and sputum) were negative. After two pulses of dexamethasone 40 mg/day for 4 days each, there was a significant clinical and laboratory improvement.
Abdominal lymphadenopathies remained stable in number and size on control CT-scan. Histologyslides were reviewed and compatible with the diagnosis of necrotic KFD. Treatment with corticosteroids was instituted for six months, with resolution of the condition and no evidence of recurrence on clinical followup.
Nine months later, the patient was hospitalized again due to asthenia, weight loss and long lasting low grade fever, with initial symptoms 1 month previous to the admission, associated with anemia (hemoglobin-9.9 g/dL), and acute kidney injury (creatinine-9.99 mg/dL). Although no peripheral lymphadenopathies were palpable, abdominal ultrasound and thoraco-abdominal-pelvic CT-scan revealed multiple mediastinal, inter-aorto-caval and lumbar-aortic lymphadenopathies, and hepatosplenomegaly.
Bone marrow cytology and immunophenotyping showed no abnormal lymphoid cells, histology evidenced changes of probable reactive etiology, without neoplastic characteristics, and karyotype did not detect chromosomal abnormalities.
Autoimmunity study, serologies for infectious agents and cultural tests were repeated, and in addition to IGRA (Interferon Gamma Release Assay) was requested, all with negative results.
Considering the previous and the current clinical history, the hypothesis of recurrence of KFD was admitted. Intravenous corticotherapy was initiated with significant clinical, imagiologic and laboratory improvement, and five months later the patient was prescribed methylprednisolone 10 mg/day, keeping KFD under control.
Later on, the patient had several hospitalizations for recurrent respiratory infections (due to Influenza A H1N1 and several nosocomial pneumonia), in the context of structural pulmonary pathology (severe emphysema of smoking etiology) and immunosuppression associated with corticosteroid therapy
Death occurred roughly a year after KFD recurrence, and about three years after initial diagnosis, following a severe respiratory infection caused by multiresistant Pseudomonas aeruginosa.
Discussion and Conclusion
KFD is a rare, benign and usually self-limiting cause of cervical lymphadenopathies and fever. It is more frequent in Asians, usually affecting young adults, typically women in their third decade of life. Rare cases have been described in the Western World.
Although etiology remains unclear, infectious agents including EBV, HHV6, parainfluenza virus, Toxoplasma gondii and Yersinia enterocolitica have been suspected to be involved [18]. Other microorganisms such as herpes simplex, CMV, varicela zoster, HIV, Rubella, Measles, Coxsackie vírus, Hepatitis A (HAV) and HBV, Influenza virus, Leptospira and Chlamydia were also related [6]. An association with SLE, mixed connective tissue disease and Still’s disease has also been postulated [19]. It is believed to be one of the self-limited causes of cervical lymphadenopathy, that is often under diagnosed [20].
Cases with unusual forms of presentation or the involvement of other lymph node groups have been described, however, generalized lymphadenopathy is uncommon [2,4,21]. Other less commonly reported presentations include fatigue, skin rash leucopenia and atypical lymphocytes on peripheral blood smear, liver dysfunction, bone marrow involvement and hepatosplenomegaly [16]. On the other hand, thrombocytopenia is a rare finding [2].
Sporadic cases of Kikuchi’s disease have been reported in Portugal [22]. This reported case describes a patient with an unusual presentation, characterized by a caucasian male in the sixth decade of life, who evidenced anemia and the simultaneous involvement of multiple lymph nodes within several locations (submandibular, cervical, inguinal, mediastinal and lumbar-aortic). Moreover, this form of presentation was associated with other less common symptoms, such as marked weight loss, asthenia, tiredness, thrombocytopenia and hepatosplenomegaly, suggesting the diagnosis of lymphoproliferative diseas
Differential diagnosis is particularly important because clinical presentation is often in distinguishable from other conditions, such as lymphoma, tuberculosis, sarcoidosis, SLE, viral infections or metastatic diseases, which have a less benign course and require more specific and targeted drug therapy. In this patient, several investigations were made towards other etiologies (infectious agents, autoimmunity and tumoral markers), all of them negative.
Although laboratory findings are usually normal, abnormalities may include leukopenia, anemia, and increase of sedimentation rate, C-reactive protein, lactate dehydrogenase and liver function enzymes [4,23]. However, the confirmation of KFD diagnosis is histopathological, requiring excisional biopsy of the involved lymph nodes. This disorder is characterized by destruction of lymph node parenchyma, and typical findings include patchy areas of coagulative necrosis in the cortical and paracortical areas of enlarged lymph nodes, together with nuclear debris or extensive karyorrhexis [24]. Microscopic examination can also demonstrate paracortical foci of histiocytic infiltrate, with characteristic absence of neutrophils and eosinophils with few or no plasma cells [19]. The immunohistologic landscape of KFD is complex and characterized by increased numbers of plasmacytoid dendritic cells that frequently cluster around apoptotic/necrotic foci, increased cytotoxic T-cells, and substantial distortion of follicular dendritic cell mesh works [25]. However, in the case reported, histological examination of the cervical lymph node revealed necrotizing lymphadenopathy with severe plasmacytosis.
In most of the reported cases, the usual recovery time is 1 to 4 months after the onset of symptoms [2,20,21,24]. Although the majority of patients has a self-limited clinical course, about 3% to 4% of patients experience recurrent episodes [14,16,20,24], developed mainly over a period of 2 to 12 years [14], but rarely as late as 19 years after initial diagnosis (one case described) [24].
In this case, two years after initial presentation the diagnosis of relapse of KFD was admitted, taking into account the presence of isolated intrabdominal lymphadenopathies, the similarity with the initial symptoms and the favourable response to corticotherapy. According to the literature, fever, fatigue, extranodal involvement and long symptomatic duration were some important predictive factors identified for recurrent KFD [14,18] and viral infections including EBV, Parvovírus B19 and HHV8 have been hypothesized to be among the triggers for KFD relapse [16]. Although the reported patient evidenced symptoms associated with recurrent disease, no specific factor of recurrence has been identified in this case
As it is a self-limited disease, most cases are controlled with supportive therapy (NSAIDs and analgesics) to relieve pain or fever, but in more severe conditions, corticosteroid therapy may be necessary and is the treatment of choice [15]. Patients with relapsing disease, or a more severe clinical course, might benefit from a prolonged corticosteroid therapy [15
In this case, due to the atypical presentation and the severity of the symptoms, there was a need for corticosteroid therapy, both in the initial episode and in the recurrence phase. The immunosuppression caused by the need for corticosteroid therapy, associated with the patient delicate structural lung disease and the difficulty to control the disorder, conditioned several hospitalizations for respiratory infection during clinical course. The patient died following several complications due to respiratory infection.
With this report, the authors intend to alert to the diagnostic difficulty of a rare cause of lymphadenopathies in the Western world, since it is not obvious, nor immediate, requiring the exclusion of other causes that are much more frequent and clinically severe, whose treatment differs significantly and is more specific. Early and reliable diagnosis of the disease using imaging technologies and laboratory tests (including diagnostic histology) and appropriate management of the symptoms are crucial to avoid misdiagnosis and mistreatment.
Declarations
Acknowledgements: Not applicable
Ethics approval and consent to participate: The authors declare that the procedures followed were in accordance with the regulations established by the heads of the Clinical and Ethical Research Commission and in accordance with the Declaration of Helsinki of the World Medical Association.
Confidentiality of the data and consent for publication: The authors declare to have followed their work center protocols on publishing patient data.
Availability of data and materials: The data that support this case report is based on clinical records.
Competing interests: The authors claim no competing financial or intellectual conflicts of interest in the preparation and submission of this manuscript.
Funding: Not applicable.
Authors´Contributions: JH, APA: drafted and wrote the manuscript. FM, FM, PSS, MC, ABR, CS, SM, FM, JV: Analyzed the data and revised the paper. MC: provided the histological images. All authors approved the final manuscript.
References
- Nair IR, et al. Clinicopathologic spectrum of necrotizing lymphadenitis. Indian J Pathol Microbiol. 2020; 63: 60-
- Ranabhat S, et al. An uncommon presentation of Kikuchi Fujimoto disease: A case report with literature review. BMC Res Notes. 2015; 8: 478.
- Sousa AeA, et al. Kikuchi-Fujimoto disease: three case reports. Sao Paulo Med J. 2010; 128: 232-235.
- Alosaimi S, et al. A Case of Kikuchi-Fujimoto Disease Associated with Erosive Lichen Planus. Cureus. 2020; 12: e7312.
- Shabana M, Warnack W. An atypical neurologic complication of Kikuchi-Fujimoto Disease. Neurol Neuroimmunol Neuroinflamm. 2020; 7.
- Kido H, et al. Kikuchi-Fujimoto disease (histiocytic necrotizing lymphadenitis) with atypical encephalitis and painful testitis: A case report. BMC Neurol. 2017; 17: 22.
- Sah SK, et al. Recurrent Kikuchi-Fujimoto disease: Case report. Br J Oral Maxillofac Surg. 2007; 45: 231-233.
- Cunha BA, et al. Fever of Unknown Origin (FUO) caused by Kikuchi’s disease mimicking lymphoma. Heart Lung. 2009; 38: 450- 456.
- Danai MM, et al. A Rare Pediatric Case of Persistent Kikuchi-Fujimoto Disease and Underlying Systemic Lupus Erythematosus. J Clin Rheumatol. 2020.
- Batton E, et al. Kikuchi-Fujimoto Disease in Children: An Important Diagnostic Consideration for Cervical Lymphadenitis. Pediatr Ann. 2019; 48: e406-e411.
- Xu S, Sun W, Liu J. Kikuchi-Fujimoto disease: A case report and the evaluation of diagnostic procedures. BMC Oral Health. 2019; 19: 223.
- Sharma K, Otieno F, Shah R, et al. Case Report of Kikuchi-Fujimoto Disease from Sub-Saharan Africa: An Important Mimic of Tuberculous Lymphadenitis. Case Rep Med. 2020; 2020: 4385286.
- Canadas S, et al. Cervical Lymphadenopathy in Two Young Women The Same Rare Diagnosis with Different Presentations. Eur J Case Rep Intern Med. 2020; 7: 001516.
- Bi L, et al. Recurrence of histiocytic necrotizing lymphadenitis: A case report and literature review. Exp Ther Med. 2014; 7: 1167- 1169
- Gerwig U, Weidmann RG, Lindner G. Relapsing Kikuchi-Fujimoto Disease Requiring Prolonged Steroid Therapy. Case Rep Emerg Med. 2019; 2019: 6405687.
- Erfanian Taghvaei, MR, et al. A Case of Recurrent Kikuchi-Fujimoto Disease. Jundishapur J Microbiol. 2015; 8: e25654.
- Honda F, et al. Recurrent Kikuchi-Fujimoto Disease Successfully Treated by the Concomitant Use of Hydroxychloroquine and Corticosteroids. Intern Med. 2017; 56: 3373-3377.
- Song JY, et al. Clinical outcome and predictive factors of recurrence among patients with Kikuchi’s disease. International Journal of Infectious Diseases. 2009; 13: 322-326.
- Srikantharajah M, et al. Kikuchi-Fujimoto disease: A rare but important differential diagnosis for lymphadenopathy. BMJ Case Rep. 2014; 2014.
- Alam H, et al. Kikuchi disease. J Pak Med Assoc. 2015; 65: 1349- 1350.
- Dalugama C, IB Gawarammana, Fever with lymphadenopathy - Kikuchi Fujimoto disease, a great masquerader: A case report. J Med Case Rep. 2017; 11: 349.
- Berger S. Infectious diseases of Portugal. 2020; Gideon E-books Series.
- Perry AM, SM Choi, Kikuchi-Fujimoto Disease: A Review. Arch Pathol Lab Med. 2018. 142: 1341-1346.
- Mrówka Kata K, et al. Kikuchi-Fujimoto disease as a rare cause of lymphadenopathy- two cases report and review of current literature. Otolaryngol Pol. 2013; 67: 1-5.
- Sukswai N, et al. Immunopathology of Kikuchi Fujimoto Disease: A reappraisal using novel immunohistochemistry combinations. Histopathology. 2019.