
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Not all lymphadenopathy is lymphoma: A case report of Rosai-Dorfman disease
Chloe Weidenbaum*; Akanksha Kushwah; Sunny S Cai
University of Tennessee Health Science Center, 2000 Church Street, Nashville, TN 37236, USA.
*Corresponding Author : Chloe Weidenbaum
University of Tennessee Health Science Center,
2000 Church Street, Nashville, TN 37236, USA.
Email: : chloe.weidenbaum@ascension-external.org
ORCID ID: 0000-0003-4246-9192
Received : May 30, 2022
Accepted : Jun 29, 2022
Published : Jul 06, 2022
Archived : www.jcimcr.org
Copyright : © Weidenbaum C (2022).
Abstract
Background: Rosai-Dorfman Disease (RDD) is a rare non-Langerhans cell histiocytic disorder most commonly presenting as bilateral cervical lymphadenopathy in children and young adults. Although a heterogeneous entity with a range of clinical phenotypes, here we describe a presentation at one of the oldest ages found to date in the available literature and its spontaneous resolution in the absence of treatment.
Case presentation: A 75-year-old man presented with new-onset right-sided cervical lymphadenopathy and fever, weight loss, fatigue, and anorexia. Physical exam revealed cervical and axillary lymphadenopathy. CT chest/abdomen/pelvis showed extensive adenopathy in the axillary regions, groin, mediastinum, retroperitoneum, splenic hilum, gastrohepatic ligament and porta hepatis, and celiac chains. Left axillary lymph node excisional biopsy revealed sinus histiocytosis with massive lymphadenopathy, consistent with a diagnosis of RDD. Flow cytometry showed mixed polytypic B-cell and T-cell subsets with polyclonal plasma cells. Immunofixation was negative for monoclonal immunoglobulins. On seeing a hematologist one month later, the patient’s symptoms had resolved and he had gained 10+ pounds.
Discussion: Rosai-Dorfman Disease is a rare non–Langerhans cell histiocytosis most often seen in children and young adults, although it has been reported up to age 79 years. It typically presents with markedly enlarged, non-tender cervical lymphadenopathy; however, other nodal and extranodal sites can be involved. Due to the rarity of the disease there is a lack of robust data regarding specific treatment modalities, although consensus guidelines were published in 2018 recommending various treatment options, including corticosteroids, chemotherapy, radiation, and surgery. Spontaneous resolution may be observed but can take many months to years.
Keywords: sRosai-Dorfman disease; Sinus histiocytosis with massive lymphadenopathy; Non–langerhans cell histiocytosis.
Abbreviations: CKD: Chronic Kidney Disease; PET: Positron Emission Tomography; RDD: Rosai-Dorfman Disease.
Citation: Weidenbaum C, Kushwah A, Cai SS. Not all lymphadenopathy is lymphoma: A case report of Rosai-Dorfman disease. J Clin Images Med Case Rep. 2022; 3(7): 1930.
Background
Rosai-Dorfman Disease (RDD) is a rare non-Langerhans cell histiocytic disorder most often presenting as bilateral cervical lymphadenopathy in children and young adults (mean age, 20.6 years), although it has been reported up to age 79 years [1,3]. It typically presents with markedly enlarged, non-tender cervical lymphadenopathy; however, other nodal and extranodal sites can be involved [2,4,5].
Laboratory evaluation typically shows leukocytosis, polyclonal hypergammaglobulinemia, normocytic anemia, and elevated ESR/CRP. These cells are S100+, CD 68+, and CD1a-, and demonstrate variable frequency of emperipolesis (the presence of one cell within the cytoplasm of another, a hallmark of RDD). A thorough work-up is required, as RDD can occur in isolation or in association with autoimmune or malignant diseases. Spontaneous resolution may be observed but can take many months if not years [5]. Due to the rarity of this disease, there is a lack of robust data regarding specific treatment modalities, although consensus guidelines were 4 published in 2018 recommending various treatment options, including corticosteroids, chemotherapy, radiation, and surgery [2].
Although a heterogeneous entity with a range of clinical phenotypes, here we describe a presentation of RDD at one of the oldest ages found to date in the available literature and its spontaneous resolution in the absence of pharmacologic treatment.
Case presentation
A 75-year-old man with a history of Chronic Kidney Disease (CKD) and anemia presented with acute-onset right-sided cervical lymphadenopathy and systemic symptoms of fever, weight loss of 35 pounds within 4 weeks, fatigue, and anorexia. Physical exam revealed cervical and axillary lymphadenopathy. Initial lab work was notable for leukocytosis with neutrophilic predominance, hyponatremia, hemoglobin 8.1, ferritin 825, and iron 26.4 g/dL.
Peripheral blood smear showed leukocytosis but was negative for blasts or atypical cells. CT scan of the chest, abdomen, and pelvis without contrast showed extensive adenopathy in the axillary regions, groin, mediastinum, retroperitoneum, splenic hilum, gastrohepatic ligament and porta hepatis, and the celiac chains. The diagnosis was established by left axillary lymph node excisional biopsy, which revealed sinus histiocytosis with massive lymphadenopathy, consistent with a diagnosis of RDD (Figures 1-4).
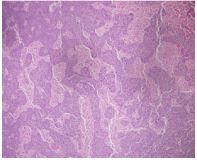
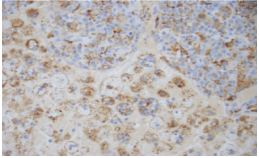
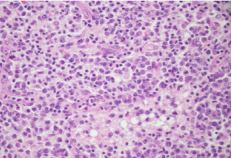
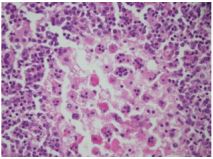
Fresh tissue analysis by flow cytometry showed mixed polytypic B-cell and T-cell subsets with polyclonal plasma cells. Immunofixation was negative for monoclonal immunoglobulins. At this point the patient was referred to a hematologist. On seeing the hematologist one month later, the patient’s symptoms had resolved and he had gained 10+ pounds. Labs at that visit resolution of the leukocytosis, persistent normocytic anemia with a hemoglobin of 7.3 (thought to be due to his CKD), and a total protein of 9.4. Unfortunately, he declined bone marrow biopsy and Positron Emission Tomography (PET) scan to evaluate for coexisting disease, including lymphoma.
Discussion
Rosai-Dorfman Disease is a rare non–Langerhans cell histiocytosis most often seen in children and young adults, although it has been reported up to age 79 years. It typically presents with markedly enlarged, non-tender cervical lymphadenopathy; however, other nodal and extranodal sites can be involved. Due to its rarity, it poses a diagnostic and therapeutic challenge. In this case, RDD was diagnosed at one of the oldest ages found to date in the available literature and demonstrates the potential for resolution of symptoms in the absence of pharmacologic treatment.
There is a lack of robust data regarding specific treatment modalities, although consensus guidelines were published in 2018 recommending various treatment options, including corticosteroids, chemotherapy, radiation, and surgery. Spontaneous resolution may be observed but can take many months to years. Although this patient’s presenting symptoms were fairly nonspecific and are not uncommon, such symptoms rarely lead to such a diagnosis. This highlights the importance of the clinician maintaining a broad differential for lymphadenopathy.
Declarations
Consent: Verbal and written consent for publication were obtained from the patient. The authors declare that they have no conflicts of interest.
Acknowledgements: We would like to thank Dr. Habib Doss for his support in the writing of this manuscript.
References
- Bruce Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: An overview. J Clin Pathol. 2020; 73: 697-705.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018; 131: 2877.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020; 105: 348- 357.
- McAlister WH, Herman T, Dehner LP. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Pediatr Radiol. 1990; 20: 425.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Review of the enstity. Semin Diagn Pathol. 1990; 7: 19