
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
SNORD118 negative Labrune syndrome
Matheus Alves da Silva¹*; Alex Machado Baeta¹; Barbara Maini de Carvalho¹; Hennan Salzedas Teixeira¹; Victor Hugo Rocha Marussi²; Carmem Lucia Penteado Lancelotti³; Viviane Cordeiro Veiga4; Feres Eduardo Aparecido Chaddad Neto5
1Department of Neurology, Beneficência Portuguesa de São Paulo, Brazil.
2Department of Neurorradiology, Beneficência Portuguesa de São Paulo, Brazil.
3Department of Neuropathology, Beneficência Portuguesa de São Paulo, Brazil.
4Department of Neurointensivism, Beneficência Portuguesa de São Paulo, Brazil.
5Department of Neurosurgery, Beneficência Portuguesa de São Paulo, Brazil.
*Corresponding Author : Matheus Alves da Silva
Department of Neurology, BP-Beneficência Portuguesa Hospital of São Paulo, São Paulo, Brazil.
Email: matheus.alves.123@outlook.com
Received : Jun 20, 2022
Accepted : Jul 22, 2022
Published : Jul 29, 2022
Archived : www.jcimcr.org
Copyright : © da Silva MA (2022).
Abstract
Objective: Labrune syndrome is a heterogeneous genetic disorder. Genetic studies may fail on detecting the SNORD118 mutations, even using exoma focusing on non coding proteins, suggesting that different mutations not yet discovered may cause the same phenotype.
Methods: We used clinical, imaging, neuropathology and genetic testing.
Results: We try to determine a mutation on a 45 years old patient with new onset headache and cerebellar ataxia which after clinical and complementary investigation, came with a diagnosis of a Labrune syndrome phenotype with a negative SNORD188 mutation.
Conclusions: The case shows us that the genetic syndrome may be caused by some other mutations not yet described.
Citation: da Silva MA, Machado Baeta A, de Carvalho BM, Teixeira HS, Rocha Marussi VH, et al. SNORD118 negative Labrune syndrome. J Clin Images Med Case Rep. 2022; 3(7): 1976.
Introduction
Labrune et al originally reported three cases of children with a large range of clinical manifestations, involving pyramidal, sensory and cerebellar signs. Imaging showed parenchymal cysts, brain calcifications and abnormal sign on white matter [1].
The diagnostic approach to genetic diseases is challenging. It need a strict correlation between the clinical manifestations, laboratory, radiologic findings and complementary methods, like cytogenetic, biochemical and molecular testing. On 2016, Jenkinson et al. discovered a mutation on SNORD118 (possibly critical for maturation of ribosomal RNAs) causing Labrune syndrome [2]. Even with this discover, some genetic tests may fail on detect the mutation, mainly when focusing on protein coding genes [3].
In this article, we describe a case of adult-onset clinical manifestations and radiological findings compatible with Labrune syndrome diagnosis associated with a negative exoma.
Case report
A 45 years old patient, from São Paulo, Brazil, presented to the emergency department complaining a sub-acute onset headache, nausea and ataxic episodes. On the physical examination, he had no cognitive or sensitive deficits, but the neurologic examination showed a bilateral dysmetria and gait ataxia. A brain magnetic resonance imaging showed a supratentorial and infratentorial leukoencephalopaty, multiple cerebral and cerebellar cysts and calcifications with one of them compressing the anterior brain stem and the cerebellar parenchyma, which caused cerebral edema and obstructive hydrocephalus. The finding images were compatible with the literature description of adult-onset leukoencephalopathy with intracranial calcifications and cysts (Labrune syndrome).
The patient had a descompressive surgery of the cyst causing the hydrocephalus, and the material’s histopathologycal analysis showed a degenerative process involving all cerebellar tissue associated with coagulative necrosis, dystrophic microcalcifications and perivascular hialinizations that lead to substitution of extensive neuropil, like the original histological description of the syndrome [1]. Following the procediment, we made a whole-exome sequencing and mutations showed no mutations on SNORD118. The ophthalmic examination made by an ophthalmologist and a neurologist revealed no retinal lesions, so Cerebrospinal Microangiopathy with Calcifications and Cysts (CRMCC), an important differential diagnosis with a different mutation, was an unlikely diagnosis. The patient had no other systemic manifestations.
One year after, the patient returned in the outpatient setting showing no neurologic deficits. He is asymptomatic since.
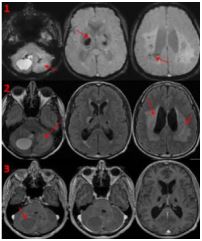
Genetic studies
Whole-Exome Sequencing (WES) was performed using Illumina technology. Sequence alignment and variants identification were made using bioinformatics protocols, using as a reference the GTCh37 version of the human genome.
99.8% of the gene targeting were read at least 10 times, with a median of 115 reads each. The analysis generated 64.816.129 sequences. The gene was entirely sequenced by Sanger technique.
CTC1 was also a target, but there was also no mutations in the gene.
Pathologic features
The biopsy analysis revealed a degenerative and regressive process on cerebellar tissue, with areas of coagulation necrosis, dystrophic microcalcifications and perivascular hialinizations that substitutes extensive areas of the neuropil.
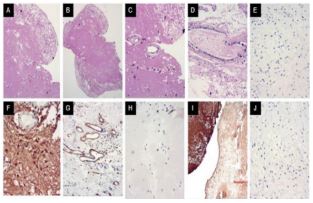
The histopathology of the cerebellar tissue may be secondary to obstructive microangiopathy leading to coagulation necrosis and, so, to the dystrophic micro calcifications.
Discussion
Leukoencephalopathy with calcifications and cysts is a genetic disorder with few literature reports since its discovery. The disease’s clinical spectrum is heterogeneous. The observation of siblings pairs raised a genetic hypothesis, confirmed on 2016 [2] by the identification of a SNORD118 mutation, which encodes the snorRNA U8 causing a ribosmopathy secondary to the biallelic mutations. These alterations causes Labrune syndrome characteristic microangiopathy [1], which is likely the causes of the clinical manifestations. A different mutation causes Coats plus syndrome, which likely causes extra neurological symptoms, like retinal disease [4].
The age of presentation is variable, ranging from the first year of life until the fifth decade of life. The mainly theory for that discrepancy is based on the funcional effects of the patient’s biallelic hypomorphic molecular lesions, which causes different rates of protein expression that leads to different phenotypes, but severity might not be predictable by the genotype alone [5]. There are many neurological manifestations associated with the syndrome, like ataxia, seizures, speech disturbance and even obstructive hydrocephalus [6], depending of the cysts localizations.
Coats plus syndrome is similar to Labrune syndrome, but this disease manifests with retinal vascular disease and a mutation on CTC1 gene, in contrast with Labrune’s SNORD118 mutation [4].
Its treatment involves supportive care, with uses of surgical approach on some cases, like when it presents with intracranial hypertension or hydrocephalus [6]. There are some reports of treatment with bevacizumab [7].
The SNORD118 mutation, like many other genetic mutations on neurologic diseases, has detection rates not so high, as suggested on past publications [4]. In addition, besides some discussion about the radiologic features, some patients with the syndrome may have a SNORD118 negative molecular testing or manifest with other mutations, like EARS2 [8].
Conclusion
We described a case with clinical manifestations, imaging and histological patterns that are compatible with Labrune syndrome with negative genetic tests focusing SNORD118. Mutation detection rates approximately 30% [3], even when not focused on protein coding genes. We suggest with the case report that next generation sequencing technologies may fail on detecting some mutations or the disease may present with different mutations.
References
- Labrune P, Lacroix C, Goutières F, et al. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: A new progressive disorder due to diffuse cerebral microangiopathy. Neurology. 1996; 46: 1297-1301.
- Jenkinson EM, Rodero MP, Kasher PR, et al. Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat Genet. 2016; 48: 1185-1192.
- Iwama K, Mizuguchi T, Takanashi JI, et al. Identification of novel SNORD118 mutations in seven patients with leukoencephalopathy with brain calcifications and cysts. Clin Genet. 2017; 92: 180-187.
- Stephani C, Pfeifenbring S, Mohr A, Stadelmann C. Late-onset leukoencephalopathy with cerebral calcifications and cysts: Case report and review of the literature. BMC Neurol. 2016; 16: 19.
- Crow YJ, Marshall H, Rice GI, et al. Leukoencephalopathy with calcifications and cysts: Genetic and phenotypic spectrum. Am J Med Genet A. 2021; 185(1):15-25.
- Shtaya A, Elmslie F, Crow Y, Hettige S. Leukoencephalopathy, Intracranial Calcifications, Cysts, and SNORD118 Mutation (Labrune Syndrome) with Obstructive Hydrocephalus. World Neurosurg. 2019; 125: 271-272.
- Martínez Matilla M, Ferre Fernández JJ, Aparisi MJ, et al. Apparent Radiological Improvement in an Infant With Labrune Syndrome Treated With Bevacizumab. Pediatr Neurol. 2020; 112: 53-55.
- McNeill N, Nasca A, Reyes A, Lemoine B, Cantarel B, et al. Functionally pathogenic EARS2 variants in vitro may not manifest a phenotype in vivo. Neurol Genet. 2017; 3: e162.