
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Mitochondrial neurogastrointestinal encephalomyopathy syndrome mimicking Guillain-Barre syndrome: A case report and literature review
Enes Tarık İnci; Tahir Kurtuluş Yoldaş
Neurology Department, Ankara Training and Research Hospital, Turkey.
*Corresponding Author : Enes Tarık İnci
Neurology Department, Ankara Training and Research Hospital, Turkey.
Phone: +905539531993, Fax: 031 595 3000;
Email: enestarikinci@gmail.com
Received : Jul 12, 2022
Accepted : Aug 05, 2022
Published : Aug 12, 2022
Archived : www.jcimcr.org
Copyright : © Tarık İnci E (2022).
Abstract
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is rare autosomal recessive multisystem disorder characterized by severe gastrointestinal dysmotility and leads to cachexia, ptosis, external ophthalmoplegia, peripheral neuropathy, and leukoencephalopathy. Due to its complex clinical findings and non-specific symptoms, diagnosis may be delayed or patients may be misdiagnosed. Multidisciplinary follow-up and treatment of patients is important because it affects many systems and does not have a specific treatment.
Keywords: Mitochondrial Neurogastrointestinal Encephalomyopathy Syndrome; Guillain-Barre Syndrome; Thymidine phosphorylase; Charcot-Marie-Tooth.
Citation: Enes Tİ, Tahir KY. Mitochondrial neurogastrointestinal encephalomyopathy syndrome mimicking Guillain-Barre syndrome: A case report and literature review. J Clin Images Med Case Rep. 2022; 3(8): 1996.
Introduction
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a neurodegenerative disease resulting from mutations in the gene encoding thymidine phosphorylase (TP). Mitochondrial myopathy with ptosis and progressive external ophthalmoplegia includes peripheral neuropathy, gastrointestinal motility disorder causing severe cachexia, and leukoencephalopathy without cognitive loss [1]. The disease begins on average in the second decade, and patients die in the fourth decade, depending on the degree of gastrointestinal involvement [2]. The first symptoms usually occur in the gastrointestinal tract, followed by symptoms of the ocular and neurological systems. Diagnosis may be difficult due to the complexity of clinical presentation [3]. A case of MNGIE presenting with subacute weakness and acute axonal loss added to demyelinating polyneuropathic involvement on electromyography is presented in our clinic.
Case report
A 19-year-old male patient presented to the emergency department with complaints of instability, loss of strength and numbness in both arms and legs that started 2 weeks ago. The general appearance of the patient was extremely cachectic. His muscles were atrophied and he had a frail appearance. He had begun to fall due to weakness and instability, which began around the age of 11. For this reason, he was frequently admitted to the hospital with extremity fractures and received medical help. From the age of 13, he had diarrhea without blood and mucus, which was 2-4 times a day. He had been followed up and treated 3 years ago for intussusception and ileus. About 6 months ago, he was examined by an ophthalmologist due to strabismus, a glasses were prescribed for him, but he could not get glasses yet. He had been admitted to the hospital many times with gastrointestinal and neurological symptoms. In his neurological examination, he had oculomotor nerve palsy in the left eye, proximal muscle strength of both lower extremities 4/5, distal muscle strength 2/5, both upper extremities proximal muscle strength 4/5, distal muscle strength 3/5. He has hypoesthesia and numbness in the glove-sock distribution in the distal of her extremities. Deep tendon reflexes were absent. The patient was able to take several steps with a single support.
Guillain-Barre syndrome and Charcot-Marie-Tooth disease were evaluated with EMG with preliminary diagnoses. EMG showed findings consistent with acute axonal loss added to demyelinating involvement (Table 1). Due to the subacute onset of the patient’s clinic and findings consistent with demyelinating polyneuropathic involvement and acute axonal loss in EMG, a 5-day intravenous immunoglobulin (IVIG) treatment at a dose of 0.4 g/kg was started with the diagnosis of Guillain-Barre syndrome.
Table 1A: Sensory NCS.
| Nerve / Sites | Rec. Site | Latency ms | Peak Ampl μV | Distance cm | Velocity m/s |
|---|---|---|---|---|---|
| L HAND - CTS | |||||
| Median Dig II | Wrist | NR | NR | 14 | NR |
| Median Palm | Wrist | NR | NR | 8 | NR |
| Ulnar Dig V | Wrist | NR | NR | 14 | NR |
| R SURAL – Lat Malleolus | |||||
| Calf | Lat Malleolus | NR | NR | ||
| L SURAL – Lat Malleolus | |||||
| Calf | Lat Malleolus | NR | NR | ||
Table 1B: Motor NCS
| Nerve / Sites | Latency ms | Ampl mV | Distance cm | Velocity m/s |
|---|---|---|---|---|
| L MEDIAN - APB | ||||
| Wrist | 4,25 | 5,6 | 5 | |
| Elbow | 11,30 | 3,4 | 21 | 29,8 |
| L ULNAR - ADM | ||||
| Wrist | 3,80 | 4,9 | ||
| B. Elbow | 10,80 | 3,8 | 23 | 32,9 |
| R COMM PERONEAL - EDB | ||||
| Ankle | NR | NR | ||
| L COMM PERONEAL - EDB | ||||
| Ankle | NR | NR | ||
| R TIBIAL(KNEE) - AH | ||||
| Ankle | NR | NR | ||
| L TIBIAL(KNEE) - AH | ||||
| Ankle | NR | NR | ||
Table 1C: Needle EMG.
| EMG Summary Table | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spontaneous | MUAP | Recruitment | ||||||||
| IA | Fib | PSW | Fasc | H.F. | Amp | Dur. | PPP | Pattern | ||
| R. TIB ANTERIOR | N | 3+ | 3+ | None | None | - | - | - | No Activity | |
| R. GASRTROCN(MED) | N | 2+ | 2+ | None | None | - | - | - | No Activity | |
| R. FIRST D INTEROSS | N | 2+ | 2+ | None | None | - | - | No Activity | ||
| R. BICEPS | N | None | None | None | None | - | - | No Activity | ||
| L. FIRST D INTEROSS | N | None | 1+ | None | None | - | - | - | No Activity | |
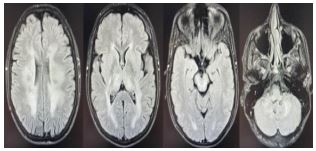
Thymidine phosphorylase gene mutation was detected in the DNA analysis sent for MNGIE due to the patient’s gastrointestinal findings, ophthalmoplegia and peripheral neuropathy. He was included in the physical therapy and exercise program in terms of pes cavus and ambulation. An ankle joint orthosis (AFO) was fitted to increase mobilization. There were no significant findings in the vasculitis panel, serum and urine immunofixation, and protein electrophoresis tests. The patient was discharged after approximately 15 days of clinical follow-up, taking a few steps alone with articulated AFO.
Conclusion
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a very rare disease caused by mutations in TYMP, the gene encoding the thymidine phosphorylase enzyme. The resulting enzyme deficiency leads to a systemic accumulation of thymidine and 2’-deoxyuridine, and eventually to mitochondrial failure due to the progressive acquisition of secondary mitochondrial DNA (mtDNA) mutations and mtDNA depletion[4].
Symptoms such as dysphagia, early satiety, nausea, vomiting and diarrhea are seen due to the degeneration of the autonomic nervous system in the digestive system. Generally, this results in increased muscle breakdown and a cachectic appearance [5]. Neurological findings such as peripheral neuropathy, mitochondrial myopathy, and progressive external ophthalmoplegia occur. Initially, the lower extremities are affected. The mean age of onset is 17.9. Patient survival is generally related to the degree of gastrointestinal involvement, and patients die at a mean age of 37.6 years as a result of cachexia, peritonitis, esophageal bleeding, intestinal rupture, or aspiration pneumonia [2].
Due to the very rarity, complex clinical presentation, and non-specific initial findings of MNGIE, patients seek medical care for many years without being properly diagnosed. Among the non-specific symptoms, 50% are gastrointestinal, 20% are ocular, and the rest are neuropathic and myopathic. Most of these symptoms begin in childhood. As a result, patients are often misdiagnosed or late, despite going through a range of specialties [3]. Reported misdiagnoses include gastrointestinal diseases such as anorexia nervosa, Chron’s disease, esophagitis and gastritis, Celiac disease, Whipple’s disease, inflammatory bowel disease, irritable bowel syndrome, and intestinal pseudo-obstruction [6-10]. Due to the progression of neurological symptoms and the emergence of new neurological findings in certain stages of the disease, some patients have been misdiagnosed as chronic inflammatory demyelinating polyneuropathy, Charcot-Marie-Tooth disease, Kearns-Sayre syndrome, and chronic progressive external ophthalmoplegia [11-13].
Patients are subjected to unnecessary treatments and surgical interventions due to misdiagnoses. As with many other rare diseases, diagnostic delays reported between 5 and 10 years in MNGIE patients are not uncommon [14]. Because the disease is a progressive neurodegenerative process, late diagnosis and treatment are associated with poor prognosis [15].
MNGIE patients often present with gastrointestinal symptoms such as diarrhea, nausea, dysphagia, pseudo-obstruction, and neurological findings such as ophthalmoplegia, peripheral neuropathy, and mitochondrial myopathy. The first symptoms usually belong to the gastrointestinal system, followed by symptoms of the ocular and neurological system. Accurate diagnosis is difficult because gastrointestinal, ocular, neuropathic and myopathic findings are rarely seen simultaneously. Diagnosis can be difficult due to the complexity of the clinical presentation, the development of new findings belonging to different systems at certain stages of the patient, and non-specific symptoms. Patients are often subjected to many examinations and treatments by various specialties until the correct diagnosis is made. When the literature is examined, it may take years to make a correct diagnosis in MNGIE, as is seen in rare diseases [3].
It is expected that promising effective treatments will be found in the near future with experimental treatment strategies such as hemodialysis and peritoneal dialysis [16], platelet infusions [14], allogeneic hematopoietic stem cell transplantation, erythrocyte encapsulated thymidine phosphorylase, and liver transplantation [1]. Therefore, early and accurate diagnosis of MNGIE patients is very important to reduce the risk of multisystemic complications and to receive effective treatment.
Declarations
Acknowledgement: None.
Conflict of Interest Statement: The authors have no conflicts of interest to declare.
Funding: None.
Ethical Approval: No approval was required for this submission.
Consent: Written informed consent was obtained from the patient for the publication of this case report. A copy is available for review by the editorial staff of this journal.
References
- Filosto M, Piccinelli S.C, Caria F, Cassarino S.G, Baldelli E, Galvagni A. et al. Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1). J. Clin. Med. 2018; 7: 389.
- Hirano M, Carelli V, De Giorgio R, Pironi L, Accarino A, Cenacchi G. et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. Inherit Metab Dis. 2021; 44(2): 376-387.
- Taanman JW, Daras M, Albrecht J, Davie CA, Mallam EA, et al. Characterization of a novel TYMP splice site mutation associated with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) Neuromuscul Disord. 2009; 19: 151–4.
- Bax BE. Mitochondrial neurogastrointestinal encephalomyopathy: approaches to diagnosis and treatment. J Transl Genet Genom. 2020 ;4: 1-16.
- Nishino I, Spinazzola A, Papadimitriou A, Hammans S, Steiner I, Hahn C.D. et al. Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol. 2000; 47: 792–800.
- Filosto M, Scarpelli M, Tonin P, Testi S, Cotelli MS, Rossi M. et al. Pitfalls in diagnosing mitochondrial neurogastrointestinal encephalomyopathy. J Inherit Metab Dis. 2011; 34: 1199–203.
- Demaria F, De Crescenzo F, Caramadre A.M, D’ Amico A, Diamanti A, Fattori F. et al. Mitochondrial neurogastrointestinal encephalomyopathy presenting as anorexia nervosa. J Adolesc Heal. 2016; 59: 729–31.
- Imperatore N, Tortora R, Gerbino N, Caporaso N, Rispo A. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) mimicking refractory celiac disease. Dig Liver Dis. 2017; 49(9): 1061-1062.
- Nagata J.M, Buckelew S.M. Mitochondrial neurogastrointestinal encephalomyopathy in the differential diagnosis of eating disorders. J Adolesc Heal. 2017; 61: 661.
- Kučerová L, Dolina J, Dastych M, Bartušek D, Honzík T, Mazanec J. et al. Mitochondrial neurogastrointestinal encephalomyopathy imitating Crohn’s disease: a rare cause of malnutrition. J Gastrointest Liver Dis. 2018; 27: 321–5.
- Bedlack R.S, Vu T, Hammans S, Sparr S.A, Myers B, Morgenlander J. et al. MNGIE neuropathy: five cases mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2004; 29: 364–8.
- Needham M, Duley J, Hammond S, Herkes GK, Hirano M, Sue C.M. Mitochondrial disease mimicking Charcot-Marie tooth disease. J Neurol Neurosurg Psychiatry. 2007; 78: 99–100.
- Prasun P, Koeberl D.D. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype in a patient with a novel heterozygous POLG mutation. J Neurol. 2014; 261: 1818–9.
- Lara M.C, Valentino M.L, Torres-Torronteras J, Hirano M, Martí R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): biochemical features and therapeutic approaches. Biosci Rep. 2007; 27: 151–63.
- Scarpelli M, Russignan A, Zombor M, Bereczki C, Zappini F, Buono R. et al. Poor outcome in a mitochondrial neurogastrointestinal encephalomyopathy patient with a novel TYMP mutation: the need for early diagnosis. Case Rep Neurol. 2012; 4: 248–53.
- Yavuz H, Özel A, Christensen M, Christensen E, Schwartz M, Elmaci M. et al. Treatment of mitochondrial neurogastrointestinal encephalomyopathy with dialysis. Arch Neurol. 2007; 64: 435–8.