
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Atypical caudal regression syndrome with lumbar agenesis, hypoplastic sacrum without sacro-iliac joints in the Eastern Democratic Republic of Congo: A case report
Paterne Mudekereza Safari1,4; Roméo Bujiriri Murhega1,2,4*; Daniel Safari Nteranya2,5; Gauthier Bahizire Murhula1,4; Alliance Wani Bisimwa1,4; Bijoux Safi Matabaro1,4; Hervé Monka Lekuya6; Ghislain Maheshe Balemba3,4; Léon-Emmanuel Mubenga Mukengeshayi1,4
1Department of Surgery, Provincial General Reference Hospital of Bukavu, Bukavu, Democratic Republic of Congo.
2Research Department, Association of Future African Neurosurgeons, Yaoundé, Cameroun.
3Department of Radiology, Provincial General Reference Hospital of Bukavu, Bukavu, Democratic Republic of Congo.
4Faculty of Medicine, Université Catholique de Bukavu, Bukavu, Democratic Republic of Congo.
5Department of Surgery, University Clinics of Bukavu, Official University of Bukavu, Bukavu, Democratic Republic of Congo.
6Department of Surgery, College of Health Sciences, Makerere University, Uganda.
*Corresponding Author : Roméo B Murhega
Avenue Wesha N23D, Kadutu, Bukavu, Democratic Republic of Congo.
Phone: +24-399-779-3770;
Email: romeobujiriri1@gmail.com
ORCID: 0000-0002-0022-6355
Received : Aug 01, 2022
Accepted : Aug 23, 2022
Published : Aug 30, 2022
Archived : www.jcimcr.org
Copyright : © Murhega RB (2022).
Abstract
Lumbosacral agenesis is a rare and complex malformation characterized by a total or partial absence of the lumbosacral vertebrae. It is often associated with other congenital malformations. Its etiology is poorly understood but factors such as gestational diabetes, genetic abnormalities and environmental factors are incriminated. We report the case of a female infant, 11 months old, who presented with lumbosacral agenesis. As our hospital was less equipped to deal with this complex malformation, we transferred the infant to another better equipped hospital.
Keywords: Lumbosacral agenesis; Atypical; Hypoplastic sacrum; Democratic republic of congo.
Citation: Safari PM, Murhega RB, Nteranya DS, Murhula GB, Bisimwa AW, et al. Atypical caudal regression syndrome with lumbar agenesis, hypoplastic sacrum without sacro-iliac joints in the Eastern Democratic Republic of Congo: A case report. J Clin Images Med Case Rep. 2022; 3(8): 2021.
Introduction
The agenesis of any segment of the lower spinal column, referred to as “Caudal Regression Syndrome” (CRS) [1], is a rare congenital malformation of the spine. This spectrum of malformations is characterized by the absence of some or the entire lumbo-sacral vertebral segment [1-3]. Its etiology remains unknown, but there are several genetic and environmental factors that are known to induce such defects during the embryonic period.
Regarding the most common form of sacral agenesis, it can be syndromic or non-syndromic, it is due to varying degrees of congenital development failures [4,5]. This condition is observed in 1 to 25,000 living births and it is clinically marked by diverses clinical manifestations dependent on the segment concerned [6,7]. An isolated lumbar agenesis with or without a sacrum is rarely reported. Recent 2 case reports published by Szumera et al. have described a case of lumbar agenesis with an eutrophic sacrum [8]. In the management of their cases, the presence of the sacrum was useful for deformity corrections with hardware.
Although some cases have been reported in the Maghrebine part of Africa [9], Very few of these congenital malformations have been reported in Sub-saharan Africa. We are reporting anatypical case of the caudal regression syndrome with lumbar agenesis disconnected from the remaining hypoplastic sacrum in the Eastern part of the Democratic Republic of Congo.
Case presentation
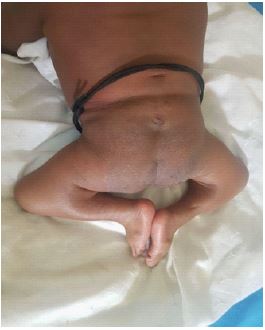
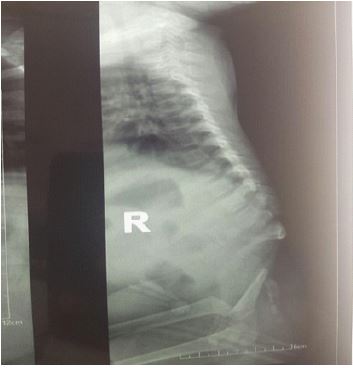
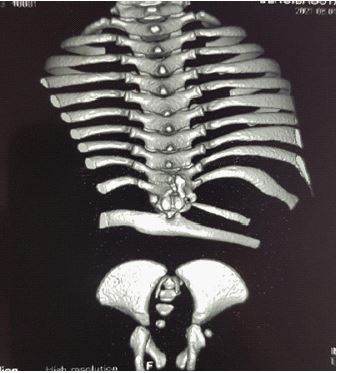
We report the case of an 11-month-old infant, female, with no particular fetal or maternal history, brought to our institution for the management of hypotonia of the lower limbs with dorsal column deformity of dorsal hyperkyphosis. Clinically we noted flexed knees with flanges in the popliteal fossae (Figure 1), anal sphincter atony and a sensation of emptiness in the lumbosacral region. The standard X-ray performed shows an absence of the lumbosacral spine (Figure 2); the abdominopelvic ultrasound performed to look for associated malformations was normal. A 3D CT scan of the whole spine showed absence of the lumbar spine disconnecting the upper segment of the thoracic spine from the remaining hypoplastic sacrum. We noted also the absence of the sacro-iliac joints bilaterally. The last floating ribs were fused with the last Thoracic Vertebra (T12). The rest of the spine and the thoracic bones appear normal (Figure 3).
As our hospital was less equipped to deal with this complex malformation, we transferred the infant to another better equipped hospital.
Discussion
CRS is a rare congenital malformation due to a defect in the intrauterine development of the caudal portion of the spine, especially of the sacrococcygeal or, sometimes, lumbosacral segment, around the fourth week of embryonic life [10]. Its incidence is 1/10000 births with an etiopathogeny that remains unknown today, despite the advancement of several hypotheses, including uncontrolled maternal diabetes during pregnancy (200 times higher risk), a vascular origin by hypoperfusion (single umbilical artery), genetic factors or toxicity (drugs) [11].
Several studies have been conducted in terms of caudal regression syndrome and many types of classifications of caudal regression syndrome have been established: Duhamel [12], Renshaw and Guille [13]. The last one has the advantage of predicting the ambulatory potential by identifying patients who will benefit from early operative treatment of the lower-extremity deformities to facilitate walking [14]. In fact in the guile classification of CRS, 2 types of patients are described: group I, which include the absence of myelomeningocele and Group II without myelomeningocele in which group there are 3 subtypes. We report a Group I case without myelomeningocele. We should notice that the CT and X rays, agenesis of the lumbar spine disconnecting the upper segment of the thoracic spine from the remaining hypoplastic sacrum and the absence of the sacro-iliac joints bilaterally. This descriptive fits more with the subtype c of group II of the Guilty‘s classification made of total agenesis of the lumbar spine; the ilia are fused together, and there is a visible gap between the most caudal intact thoracic vertebra and the pelvis [14].
Several spinal abnormalities have been associated with CRS. Among them, there are fibro lipoma, lipoma, diastematomyelia, syringomyelia, kyphosis, myelomeningocele used even in the guile’s classification of CRS [14-16]. In our case, it has just been reported dorsal kyphosis as a spinal abnormality associated. Kyphosis has been reported among the more frequent spinal abnormalities especially in the group II (with myelomeningocele) [13,17]. It has been described also a spinopelvic dissociation due to the absence of sacroiliac joint bilaterally that may lead to walking instability as described also by Zhang et al [5].
Most of the time clinical features associated with CRS are neurologic and orthopaedic ones [13]. Our case is not spared from that because we have found orthopaedic manifestations: Flexed knees with flanges in the popliteal fossae and lower limbs hypotonia. Similar cases with lower limbs hypotonia have been described in the literature [16,18]. As neurologic manifestations, and anal sphincter atony has been found like in Castillo et al [19]; but it has been described many other neurologic consequences of the CRS like bowel dysfunction, neurogenic bladder [6,7,20].
It is recognised that early fetal MRI is the gold standard in detecting CRS [21,22]; but in the Low and middle income countries ‘context patients do not attend prenatal visits and if attended they cannot have access to such imaging tools. That is why in some studies like ours CRS is diagnosed in the neonatal period in most cases, in the childhood and some exceptions in the adulthood. In our case we were able to perform only the CT scan, US and X rays that revealed the absence of the associated visceral abnormalities. That is why this case remains atypical because Genitourinary, anorectal, digestive, cardiac malformations and other extra spinal abnormalities have been found to be associated with CRS. Anorectal together with genitourinary malformations were the mostly frequent found, and especially anal imperforation was the common extra spinal manifestation [12], followed by lower limbs deformities [21]. Some cases of associated congenital heart defect [18], horseshoe kidney have been described too [7].
CRS is a complex malformation and thus requires a thorough evaluation for an effective management. The management is variable according to the form of the disease: whether the sacral agenesis is accompanied with an open dysraphism or not [9,16,22]. The MRI and the Sonographic examination are quite necessary in the investigation of the disease; the management is multidisciplinary and depend upon the degree of the defect. Management methods vary from conservative to surgery [5,9]. Spine reconstruction has proven itself to be a valuable management technique, ilium-sacrum-ilium internal fixation and fusion are the most applied techniques. Additional treatments such as pudendal neuromodulation, physiotherapy are often integrated in the treatment schemes in regards to the accompanying malformations [5,20].
Conclusion
In this work we reported the case of an infant with an atypical form of caudal regression syndrome with lumbar agenesis, hypoplastic sacrum without sacroiliac joints. The risk factors most often described in the literature were not found in our patient. Unfortunately, we could not perform genetic analysis due to lack of expertise and equipment in our facility. The diagnosis can be made early before birth during prenatal consultations. With this work, we wanted to draw the attention of the medical world to the existence of this rare malformation in the east of the Democratic Republic of Congo.
Declarations
Acknowledgment: None.
Conflicts of interest: None.
Author contributions: Paterne Mudekereza Safari, Roméo Bujiriri Murhega, Daniel Safari Nteranya, Gauthier Bahizire Murhula, Alliance Wani Bisimwa, Bijoux Safi Matabaro, ,Hervé Monka Lekuya, Ghislain Maheshe Balemba, and Léon-EmmanEmmanuel Mubenga Mukengeshayi did the design, conceptualization, and critical review of the manuscript. Paterne Mudekereza Safari, Roméo Bujiriri Murhega, Daniel Safari Nteranya, and Gauthier Bahizire Murhula wrote the first drafts of the manuscript. All authors have reviewed and approved the last version of the work.
Ethical statement: This case report received ethical clearance from the Ethical Committee of the Catholic University of Bukavu.
Consent: Written informed consent was signed by the patient’s parent before publication of this article.
References
- Zhang T, Shu S, Jing W, Gu Q, Liu Z, Sun X, et al. Sacral Agenesis: A Neglected Deformity That Increases the Incidence of Postoperative Coronal Imbalance in Congenital Lumbosacral Deformities. Glob Spine J. 2020; 2192568220970509.
- Estin D, Cohen AR. Caudal agenesis and associated caudal spinal cord malformations. Neurosurg Clin N Am. 1995; 6: 377–391.
- Bevanda K, Memidžan I, Boban Raguž A. Caudal regression syndrome (Currarino syndrome) with chromosomal mutation 9. Radiol Case Rep. 2020; 15: 1184–1188.
- Nalbandyan M, Howley MM, Cunniff CM, Romitti PA, Browne ML, et al. For the National Birth Defects Prevention Study. Descriptive and risk factor analysis of nonsyndromic sacral agenesis: National Birth Defects Prevention Study, 1997–2011. Am J Med Genet A. 2019; ajmg.a.61290.
- Zhang H, Guo H, He S, Hui H, Hao D. Sacral agenesis combined with spinopelvic dissociation. Medicine (Baltimore). 2018; 97: e12162.
- Le HK, Cardona Grau D, Chiang G. Evaluation and Long-term Management of Neurogenic Bladder in Spinal Dysraphism. 16.
- Salsi G, Bellussi F, Pilu G, Gatta AND, Youssef A. Pregnancy in a woman with sacral agenesis from prenatal counseling to delivery: A case report. J ObstetGynaecol Res. 2020; 46: 784–786.
- Szumera E, Jasiewicz B, Potaczek T. Atypical caudal regression syndrome with agenesis of lumbar spine and presence of sacrum – case report and literature review. J Spinal Cord Med. 2018; 41: 496–500.
- Doulhousne H, Roukhsi R, Elhend SB, Hammoune N, Mouhsine A, El Fikri A, et al. Imagerie des agénésies sacrées: A propos de deux observations. PAMJ Clin Med [Internet]. 2019 [cited 2021 Aug 22]; 1. Available from: https://www.clinical-medicine.panafrican-med-journal.com/content/article/1/63/full/
- Armas EMF, Paramo LM, Marmolejo MAC, Pedraza NR, ANCHEYTA MAA, et al. Caudal regression syndrome: A review for the radiologist. [Internet]. ECR 2020 EPOS. European Congress of Radiology - ECR 2020; 2020 [cited 2021 Jul 17]. Available from: https://epos.myesr.org/poster/esr/ecr2020/C-01531
- Camacho Montaño AM, Reinaldo RCA, Grajales MCC. Dorsolumbosacral agenesis: Case report and literature review. Obstet Gynecol Int J [Internet]. 2021; 12. Available from: https://medcraveonline.com/OGIJ/dorsolumbosacral-agenesis-case-report-and-literature-review.html
- Duhamel B. From the Mermaid to Anal Imperforation: The Syndrome of Caudal Regression. Arch Dis Child. 1961; 36: 152–155.
- Balioğlu MB, Akman YE, Ucpunar H, Albayrak A, Kargın D, Atıcı Y, et al. Sacral agenesis: Evaluation of accompanying pathologies in 38 cases, with analysis of long-term outcomes. Childs Nerv Syst.2016; 32: 1693–1702.
- Guille JT, Benevides R, DeAlba CC, Siriram V, Kumar SJ, et al. Lumbosacral Agenesis: A New Classification Correlating Spinal Deformity and Ambulatory Potential: J Bone Jt Surg-Am. 2002; 84: 32–38.
- Croci DM, Dalolio M, Schaeren S, Wasner MG, Mariani L, et al. Thalidomide Embryopathy as Possible Cause of Anterior Sacral Meningocele: A Case Report: Thalidomide Embriopathy as Possible Cause of Meningocele. Birth Defects Res. 2017; 109: 1390–1392.
- Mottet N, Martinovic J, Baeza C, Guimiot F, Bault JP, Aubry MC, et al. Think of the Conus Medullaris at the Time of Diagnosis of Fetal Sacral Agenesis. Fetal Diagn Ther. 2017; 42: 137–143.
- Mathews CS, Bumpass DB, Mc Cullough FL, McCarthy RE. Expansion Thoracoplasty as a Life-Saving Procedure in an Adolescent With Severe Spinal Deformity and Sacral Agenesis. Spine Deform. 2019; 7: 171–175.
- Zhang Y, Sun C, Jiang C, Zhao W, Wang W, Cao Q, et al. Prenatal diagnosis of caudal regression with heterotaxy syndrome: A mermaid with a broken heart. Echocardiography. 2019; 36: 415–418.
- Castillo J, Cristóbal L, Alonso J, Martín R, Suárez D, Martínez MA, et al. Sacral nerve stimulation lead implantation in partial sacral agenesis using intra-operative computerized tomography. Colorectal Dis. 2016; 18: O330– O333.
- Schober MS, Ching CB, Peters KM, Alpert SA. Novel Use of Pudendal Neuromodulation in a Pediatric Patient with Caudal Regression and Partial Sacral Agenesis for Refractory Bowel Bladder Dysfunction. Urology. 2016; 94: 224–226.
- Bray JJH, Crosswell S, Brown R. Congenital talipes equinovarus and congenital vertical talus secondary to sacral agenesis. BMJ Case Rep. 2017; bcr-2017-219786.
- Mottet N, Chaussy Y, Auber F, Guimiot F, Arbez Gindre F, et al. How to Explore Fetal Sacral Agenesis without Open Dysraphism: Key Prenatal Imaging and Clinical Implications. J Ultrasound Med. 2018; 37: 1807–1820.