
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
A case of PTPN11 mutation-related Noonan syndrome
Xiao Jiang1; Xiaotong Gu1; Pengqian Li1; Xinyu Zhao1; Chuchu Wan2; Tao Luo1; Haixia Liu1*
1The Second Affiliated Hospital of Dalian Medical University, Dalian, China.
2Dalian Medical University, Dalian, China.
*Corresponding Author : Haixia Liu, PhD, MD
Department of Endocrinology and Metabolism, The Second Hospital of Dalian Medical University, Dalian 116027, PR China.
Tel: +86-411-84671291, Fax: +86-0411-84672130;
Email: dllhx1017@163.com
ORCID: 0000-0002-0529-0428.
Received : Aug 01, 2022
Accepted : Aug 24, 2022
Published : Aug 31, 2022
Archived : www.jcimcr.org
Copyright : © Liu H (2022).
Abstract
Introduction: Noonan Syndrome (NS) is an autosomal dominant disorder in which parents with Noonan syndrome have a 50% chance of passing the mutation to their children, most commonly due to a mutation in the PTPN11 gene. In a clinical case, we identified a male child who showed clinical features such as short stature, congenital heart disease and a peculiar facial appearance, strongly suspecting Noonan syndrome. The purpose of this article is to report the presentation of a case of Noonan syndrome.
Methods: Blood was drawn from the child for genetic testing.
Results: A heterozygous missense variant c.215C > G: p.A72G was detected in the subject’s PTPN11 gene, which was verified by generation sequencing, indicating that the subject’s parents did not carry the variant, presumably the variant in this child was de novo or germ cell chimerism in one parent. The child was diagnosed with Noonan syndrome.
Conclusion: The diagnosis of Noonan syndrome was established in this case because of the peculiar facial features, atrial septal defect, coagulation abnormalities, and the presence of missense mutations in the PTPN11 gene by genetic testing. No mutation of this gene was found in the parents of this case, and it is presumed that the mutation in this affected child is a de novo mutation or germ cell chimerism in one parent. Therefore, the couple is still at risk of recurrence if they have another child.
Citation: Jiang X, Gu X, Li P, Zhao X, Liu H, et al. A case of PTPN11 mutation-related Noonan syndrome. J Clin Images Med Case Rep. 2022; 3(8): 2022.
Introduction
Noonan syndrome is a autosomal dominant genetic disorder, occurred in 1/1000–2500 live births [1]. Mutations in the relevant genes are the main cause in the development and occurrence of Noonan syndrome. Mutations in PTPN11 account for up to 50% of NS patients [2]. The clinical feature of Noonan syndrome was similar to RAS opathies, Aarskog syndrome/faciogenital dysplasia, Baraitser-Winter syndrome, fetal alcohol syndrome, neurofibromatosis type 1, and Turner syndrome. Microscopically, it may be easily misdiagnosis [3]. In this report, we describe this case and review the literature in order to enhance our understand of this disease, avoid misdiagnosis, conduct effective treatment and provide evidence for its clinical treatment and prognosis.
Case report
A 12-year-old boy was admitted to the Department of Endocrinology and Metabolism, The Second Hospital of Dalian Medical University, Dalian, Liaoning Province, China, with the reason of growth slowly in height for 12 years. He had an atrial septal defect at the age of 2.5 years and was treated with an atrial septal repair. The patient denied a family history of hereditary disease. No recorded of slow growth or growth a disease was found in his parents and family.
Physical examination was displayed in Table 1. The patient was 125 cm tall, weighed 23 kg, had a waist circumference of 49 cm, a hip circumference of 61 cm, a BMI of 14.7 kg/m2, an upper volume of 62.5 cm and a lower volume of 62.5 cm (Table 1).
Intelligence was normal. The patient was dentally hypoplastic with poorly aligned teeth, moderate crowding, and high arched palatal cover (Figures 1,2). Mild elbow valgus and mild corns, symmetrical subcutaneous hemorrhage with hyperpigmentation in both lower extremities. The external genitalia were smaller than those of children of the same age, and the penis was short, about 2.5 cm long.
Laboratory tests was showed in Table 2. Growth hormone stimulation test was incomplete stimulation. growth hormone stimulation test: GH: 1.49 (0 min) 1.17 (30 min) 2.38 (60 min) 7.25 (90 min) 2.14 (120 min).
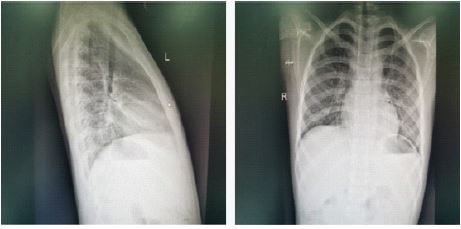
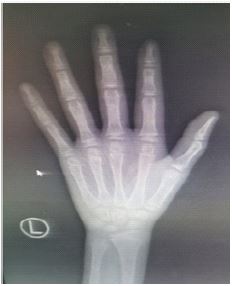
Imaging tests showed: Orthopantomogram and lateral radiographs of the chest showed that the patient had mild cervical spondylosis and chicken breasts (Figure 3). Cardiac ultrasonography showed no shunt at the atrial level and mild tricuspid regurgitation after repair of the atrial septal defect. Orthogonal DR of the left wrist bone suggests that the left wrist bone is lagging behind the actual age (Figure 4). Pituitary MRI scan showed no abnormalities in pituitary size and morphology. Enhanced examination showed small class of circular hypoenhancing areas in the pituitary gland, microadenoma cannot be excluded. Ultrasound examination of the external genitalia showed a left testicle size of 1.9*0.9 cm and a right testicle size of 2.0*0.8 cm.
Table 1: Physical tests.
| Parameter. | |
|---|---|
| T | 36.80C |
| P | 58 /min |
| R | 17 /min |
| BP | 83/50 mmHg |
| Height | 125 cm |
| Weight | 23 kg |
| Waist circumference | 49 cm |
| Hip circumference | 61 cm |
| BMI | 14.7 kg/m2 |
| Upper measurements | 62.5 cm |
| Lower volume | 62.5 cm |
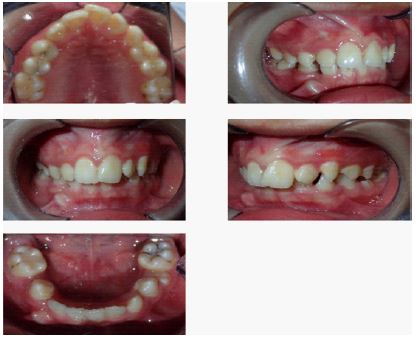
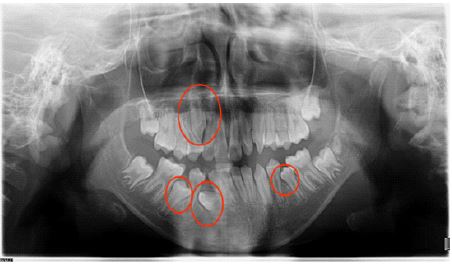
Table 2:Laboratory tests.
| TSH mIU/L | FT3 pmol/L | FT4 pmol/L | |
|---|---|---|---|
| Thyroid function testing | 1.055 | 6.27 | 18.4 |
| OGTT | Blood glucose mmol/L | Insulin mU/L | C-peptide ng/L |
|---|---|---|---|
| Fasting | 5.36 | 10.53 | 1.09 |
| 1 hour | 7.90 | 83.46 | 7.48 |
| 2 hour | 5.64 | 32.64 | 4.41 |
| 0’ | 30’ | 60‘ | 90’ | 120’ | |
|---|---|---|---|---|---|
| Growth hormone stimulation testing> | 1.49 | 1.17 | 2.38 | 7.25 | 2.14 |
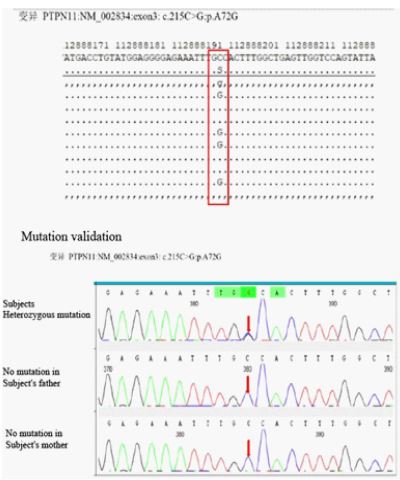

Genetic testing showed: A heterozygous missense variant c.215C > G: p.A72G was detected in the PTPN11 gene of the subject, and the results of generation sequencing showed that the subject’s parents did not carry the variant (Figure 5). The whole complete evidence of genetic testing supported the final diagnosis of Noonan syndrome. The family tree of genetics was shown in Figure 6.
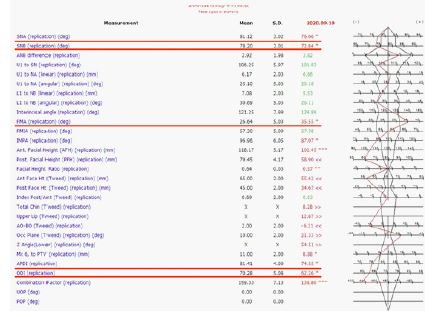
X-ray cephalometric analysis: The SNA, SNB, APOI, DOI of patient are lower than normal value. FNA are above normal range (Figure 7).
To address short stature, the patients received a dose of GH therapy (Subcutaneous injection, 5.5 IU/ day), following up with 4 weeks. For the patient’s narrow maxillary arch and relatively posterior mandibular position, the maxillary 13 eruption gap was insufficient to cause blockage. Due to the patient’s developmental potential, the patient received a two-stage treatment, in which the maxillary arch was expanded first, and after the expansion, the gap was opened with a push spring to incorporate 13 into the dentition.
Discussion
Noonan syndrome is a congenital genetic defect disorder, causing short stature, heart disease, specific appearance and other physical defects [4]. It is a autosomal dominant disorder with estimated morbidity of 1/1000–2500 live births. Mutations are the underlying cause [1,3].
The Clinical manifestations and feathers of Noonan syndrome was various and involved in multi-system & multi-organ. All patients have special facial features, including wide eye spacing, inner canthus, drooping and downward sloping eyelids [4]. Pediatric patients may also have a full forehead, a low posterior hairline, a short anterior nose, a full nasal tip, a low nasal bridge, a deep and wide nasolabial fold that reaches the upper lip, and thick lips. Though patients are born with normal stature, they appear to be short in stature and behind in age after 1 years old [3]. Thoracic deformities are the first manifestations of Noonan syndrome such as pectus excavatum or funnel chest and wide breast distance [5]. 50% to 80% of patients have congenital heart disease, including pulmonary artery constriction, hypertrophic cardiomyopathy, atrial septal defect, aortic constriction, and arteriovenous ductus arteriosus, which may present with palpitations, chest tightness, cyanosis, weakness, and dyspnea [6]. Neurological, cognitive and behavioral problems and Blood clotting disorders occurs frequently in NS patients. Female patients may have delayed transitional puberty and basically normal ovarian function and secondary sex characteristics development. In males, about half of the patients have normal testicular function, while the rest may have cryptorchidism, azoospermia, delayed puberty, hypogonadism, and renal malformation. Other signs and symptoms include elbow valgus, strabismus, eye tremor, and hepatosplenomegaly. There are also oral manifestations, consisting high palatal vault, delayed tooth eruption, jaw development [3]. The ectopic rate of Noonan syndrome is difficult to determine, as the clinical presentation of patients is variable and the diagnosis is made in many adult individuals only after the birth of an obviously affected child.
Mutations in genes are the main cause of Noonan syndrome, including PTPN11, SOS1, RAF1, RITI1, KRAS, NRAS, BRAF, MAP2K1, etc [7]. Mutations in the PTPN11 gene account for approximately 50% of Nonna syndrome [8]. PTPN11 is located on chromosome 12q24 and contains 16 exons, encoding a total of 593 amino acids [9]. Noonan syndrome with PTPN11 mutations is more likely to be familial than Noonan syndrome without PTPN11 mutations and is associated with pulmonary stenosis or atrial septal defect, bleeding disorders, and juvenile monocytic leukemia, and is negatively associated with hypertrophic cardiomyopathy and aortic coarctation.
Noonan syndrome is difficult to identify from other diseases. It is prone to misdiagnosis because of its complex clinical symptoms. The characteristic manifestations of Noonan syndrome include peculiar facial features, short stature, thoracic deformities, congenital heart disease and coagulation disorders. There is no cure for the disease, only regular follow-up is the only way to detect abnormalities and provide symptomatic treatment. The prognosis is mainly related to the severity of the cardiac lesion. In this case, we recommended growth hormone treatment for the patient’s short stature, which is effective and safe in the long term. In view of the narrow maxillary arch, the relatively posterior position of the lower jaw, and the lack of space for maxillary 13 eruption resulting in blockage, the patient still has developmental potential. The diagnosis of Noonan syndrome was established because of the peculiar facial features, atrial septal defect, coagulation abnormalities, and the presence of a missense mutation in the PTPN11 gene by genetic testing. In this case, no mutation was found in the parents, and it is assumed that the mutation in this child is either de novo or a germ cell chimerism in one of the parents. Therefore, the couple is still at risk if they have second child. After 2 months follow-up, the child was grew 3 cm in height.
Conclusion
In this case, no mutation was found in the parents, and it is assumed that the mutation in this child is either de novo or a germ cell chimerism in one of the parents. Therefore, the couple is still at risk if they have second child. After 2 months follow-up, the child was grew 3 cm in height. We report this case aims to conducted systematic review of Noonan syndrome, enhancing the capability of the diagnosis and treatment of the disease. With the natural history largely unknown, it is necessary to find the pathogenesis and mechanism of Noonan syndrome and provide new ideas for the treatment and prognosis of this disease.
Declarations
Statement of conflict of interest: The authors declare that they have no competing interests.
Consent for publication: Not applicable.
Acknowledgement: Thanks for the approval of the Second Hospital of Dalian Medical University.
Data availability: The metabolomic datasets generated during the current study are available from the corresponding author on request.
References
- Yart A, Edouard T. Noonan syndrome: An update on growth and development. Curr Opin Endocrinol Diabetes Obes. 2018; 25: 67-73.
- Tafazoli A, Eshraghi P, Koleti ZK, Abbaszadegan M. Noonan syndrome - a new survey. Arch Med Sci. 2017; 13: 215-222.
- Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics. 2010; 126: 746–759.
- Digilio M, Marino B. Clinical manifestations of Noonan syndrome. Images Paediatr Cardiol. 2001; 3: 19-30.
- Tang X, Chen Z, Shen X, Xie T, Wang X, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC Pediatr. 2022; 22: 142.
- Linglart L, Gelb BD. Congenital heart defects in Noonan syndrome: Diagnosis, management, and treatment. Am J Med Genet C Semin Med Genet. 2020; 184: 73-80.
- Zenker M, Edouard T, Blair JC, Cappa M. Noonan syndrome: Improving recognition and diagnosis. Arch Dis Child. 2022: Archdischild-2021-322858.
- Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013; 381: 333-342.
- Digilio M, Marino B. Clinical manifestations of Noonan syndrome. Images Paediatr Cardiol. 2001; 3: 19-30.