
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
A case report of hyperhomocysteinemia type II
Yongling Meng1; Guofang Xue2*; Jiying Zheng2
1The Second Clinical Medical College of Shanxi Medical University, Taiyuan 030001, China.
2Neurology Department, The Second Affiliated Hospital of Shanxi Medical University, Taiyuan 030001, China.
*Corresponding Author : Guofang Xue
Department of Neurology, The Second Affiliated Hospital of Shanxi Medical University, Taiyuan 030001, China.
Email: xueguofangty@163.com
Received : Aug 22, 2022
Accepted : Sep 12, 2022
Published : Sep 19, 2022
Archived : www.jcimcr.org
Copyright : © Xue G (2022).
Abstract
Hyperhomocysteinemia (HHcy) has been recognized as a risk factor of several chronic disease. It is noteworthy that HHcy prevalence appeared to be rising in recent years. Many patients have good curative effect after treatment, but some HHcy caused by gene mutations remains difficult to treat. Here, we present a case of hyperhomocysteinemia type II with poor effect of conventional treatment. We report it in order to provide a basis for follow-up studies.
Keywords: Hyperhomocysteinemia type II; Gene mutation; MTHFR; Treatment.
Citation: Meng Y, Xue G, Zheng J. A case report of hyperhomocysteinemia type II. J Clin Images Med Case Rep. 2022; 3(9): 2060.
Introduction
Hyperhomocysteinemia is a genetic metabolic disease, which can affect all organs and systems of the body. There are many subtypes of hyperhomocysteinemia, and each phenotype has its own special onset pattern, clinical features and associated genes. Among them, refractory hyperhomocysteinemia type II in adolescents is rarely reported. A case sent to our institution, with diagnosis of hyperhomocysteinemia type II, is described.
Case presentation
The proband is a 19-year-old man. Since 2016, his athletic ability has gradually decreased, manifested as abnormal gait and decreased muscle endurance. In March 2020, the upper limbs suddenly trembled. There were three times in one month, one of which fell to the ground, disturbance of consciousness for about two minutes, accompanied by tooth closure, salivation and stiff limbs. The disease worsened in September 2020 with specific symptoms of reduced activity, slow response and frequent limb shaking. Drugs such as gabapentin, memantine hydrochloride, flupentixol melitracen and benzhexol were taken before hospitalization, but the effect was bad. The patient had a history of intrauterine hypoxia, and his academic performance was worse than that of his peers. Parents are in good health and not close relatives. His sisters are healthy. And there is no similar disease in the family.
Physical examination: The patient’s speech was not fluent, and his advanced intelligence was significantly reduced, including memory, calculability, orientation, understanding and judgment. His eyes’ movement was restricted. The muscle tension of limbs was increased, the tendon reflex were hyperactive, the bilateral babinski sign was negative, and his gait was spastic. His body gave off a peculiar unpleasant smell. At admission, the Mini-Mental State Examination (MMSE) score was 13 points, and the Montreal Cognitive Assessment Scale (MoCA) score was 5 points.
Laboratory examination: At admission, he had significantly increased homocysteine, 186.00 μmol/L (range< 15.00 μmol/L). Folate decreased to 5.48 nmol/L (range 13.4-56.20 nmol/L), serum vitamin B12 decreased to 97.00 pmol/L (range 133.00-675.00 pmol/L). There were no significant abnormalities in the detection of lactic acid, pyruvate, amino acids and acylcarnitine.
Imaging and electrophysiological examination: MRI showed patchy long T1 and long T2 abnormal signals in bilateral lateral ventricles and frontal lobes, and high signal in T2 FLAIR. Magnetic Resonance Spectroscopy (MRS) combined with MRI plain scan showed that right frontal lobe lesions were demyelinating lesions. And there was no abnormality in cervical and thoracic MRI. Electroencephalogram (EEG) showed abnormal video electroencephalogram. Electromyography (EMG) showed multiple, mild to moderate peripheral nerve damage and nerve center damage.
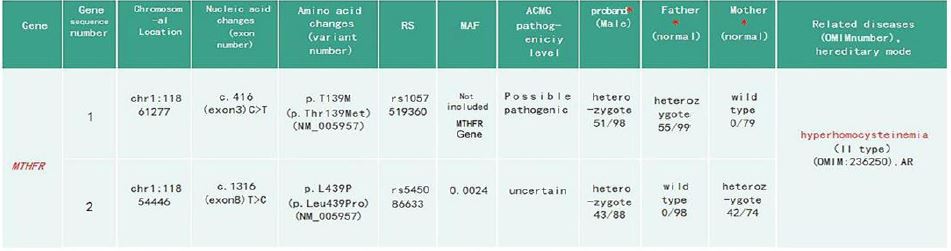
*represents the carrying state of the mutation site and the ratio of the mutation depth to the total depth.
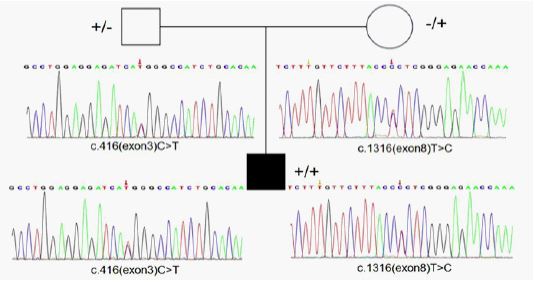
Genetic studies: Whole-Exome Sequencing (WES) showed that there were c.416 (exon 3) C>T and c.1316 (exon 8) T>C mutations in the MTHFR gene of the proband, both of which were missense mutations. Sanger sequencing showed that c.416 (exon 3) C>T originated from his father, and c.1316 (exon 8) T>C originated from his mother (Figure 1).
Treatment: The patient was diagnosed with hyperhomocysteinemia type II, and administered oral folic acid (7.5 mg/day), vitamin B1 (30 mg/day), vitamin B6 (10 mg/day), vitamin B2 (15 mg/day), mecobalamin (500 μg/day), betaine (3 g/day), levocarnitine (0.5 g/day), and was injected cobalt adenosine (twice a week). After treatment, the patient’s speech fluency, calculation ability and spastic gait tended to improve, the limbs no longer trembled, and the homocysteine level decreased to 102.1 μmol/L. And MMSE score was 17 points and MoCA score was 13 points. However, the patient is still spastic gait, the advanced intelligence is still low, and the homocysteine level is still high.
Discussion
Hyperhomocysteinemia type II is a genetic disease that is relatively rare in adolescent/adult onset. The clinical spectrum of the disease is heterogeneous. There are many neurological manifestations associated with the syndrome, like cognitive decline, behavioral abnormalities, seizures, gait disorders, encephalopathy, thrombotic events, myasthenia, motor disorders and paresthesias [1,2]. Hyperhomocysteinemia type II is due to MTHFR gene mutation. The full-length cDNA of MTHFR gene is 2.2 kb, located on chromosome 1p36.3. It contains 11 exons and 10 introns [3]. The common mutation of MTHFR is the C677T site in exon 4, which causes cytosine (C) to be replaced by thymine (T), and changes the amino acid encoded by MTHFR from valine to alanine. Thus three genotypes C/C, C/T and T/T are generated. The mutation reduce the thermal stability of MTHFR, resulting in the decrease of MTHFR enzymatic activity at temperatures above 37°C [4]. Patients with severe MTHFR deficiency have enzyme activity in the range of 0-34% [5]. The age of presentation is variable. This may be due to the functional effect of molecular damage in patients with double allele subtypes, but its severity may not be independently predicted by genotype [5]. Clinical studies suggest that nutritional factors and lifestyle (excessive or insufficient intake of folic acid, vitamin B6, vitamin B12, drinking and smoking) are as important as genetic environment for HHcy. And the interaction between two conditions is important for the understanding of HHcy [6,7]. In other words, the ultimate consequence of total hcy is the sum of interaction between genetic and environmental conditions including hcy excretion. It seems important to remember that severe HHcy is known to be not only found in early life due to genetic defect but also produced during adult ages from long period of nutritional inadequacy. This implies that the severity of HHcy is rather determined by the combination of both the magnitude and duration of metabolic deficiency [8].
Exon sequencing of the MTHFR gene was ordered to make a genetic diagnosis. Surprisingly, the sequencing identified very rare compound heterozygous mutations of the proband’s MTHFR gene. The two missense mutations were c.416 C>T (nucleotide mutation from C to T at codon 416) and c.1316 T>C (nucleotide mutation from T to C at codon 1316). As a result, threonine at position 139 was mutated to methionine and leucine at position 439 was mutated to proline. According to the guidelines of the American College Of Medical Genetics and Genomics (ACMG) in 2015, c.416 C>T is a possible pathogenic factor, and c.1316 T>C is an indeterminate mutation. So far, c. 1316 T > C, a mutation with unknown influence, has also been reported [9]. Combined with the cases, all of genotyping, function analysis, clinical diagnosis, and the effect of therapy leads to the conclusion that c.1316 T>c is a detrimental mutation instead of a benign SNP [9]. The change of codons lead to abnormal transcription and translation, affecting the activity of the enzyme and reducing the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate. It further affects the subsequent metabolism, resulting in the increase of homocysteinemia level.
The biochemical therapeutic goals in MTHFR deficiency are aimed at reducing plasma homocysteine level. Healthy lifestyle, such as smoking cessation, alcohol restriction, reasonable diet, and increased exercise, can help reduce the level of hcy. In terms of drug, we need to use the metabolic therapeutic strategies that aim at (I) enhancing methionine synthesis (using B vitamins), (III) bypassing methionine synthase using betaine (cofactor of another enzyme involved in homocysteine remethylation), and (III) supplementing methionine if needed [1,10,11]. High doses of betaine are the mainstay of therapy for patients with MTHFR deficiency. The beneficial effect of betaine is mediated through betaine-homocysteine methyltransferase with the use of an alternate methyl donor for remethylation of homocysteine to methionine. Indeed, betaine supplementation has been shown to decrease homocysteine levels and normalize methionine levels in patients with MTHFR deficiency [10,11]. Folic acid and supplementation with vitamins B6 and B12 also play key roles in converting homocysteine into methionine. Some patients improved upon B9 supplementation [1]. In addition, active folate can be used instead of ordinary folate, and vitamin B12 can be injected intramuscularly for patients with severe or complicated anemia. As expected in our case, the supplementation with betaine and multi-vitamins resulted in a decrease in the level of total plasma homocysteine, that were associated with the improvement of psychosis and the recovery of cognitive and motor functions.
Conclusion
The clinical manifestations of hyperhomocysteinemia type II are not typical, and the diagnosis is often misdiagnosed and delayed. So blood biochemical tests should be performed early to find clues in these patients. If the homocysteine level is moderate or high, gene detection should be carried out. Hyperhomocysteinemia caused by genetic variation has poor response to conventional treatment. Effective treatments, basic research and large-scale clinical trials are urgently needed to provide guidance.
Declarations
Conflict of interest: The authors have no conflicts of interest or competing interests to disclose.
References
- Gales A, Masingue M, Millecamps S, et al. Adolescence/adult onset MTHFR deficiency may manifest as isolated and treatable distinct neuro-psychiatric syndromes. Orphanet J Rare Dis. 2018; 13: 29.
- Marelli C, Lavigne C, Stepien KM, et al. Clinical and molecular characterization of adult patients with late-onset MTHFR deficiency. J Inherit Metab Dis. 2021; 44: 777-786.
- Liew SC, Gupta ED. Methylenetetrahydrofolate Reductase (MTHFR) C677T polymorphism: epidemiology, metabolism and the associated diseases. Eur J Med Genet. 2015; 58: 1-10.
- Meleady R, Ueland PM, Blom H, et al. Thermolabile methylenetetrahydrofolate reductase, homocysteine, and cardiovascular disease risk: the European Concerted Action Project. Am J ClinNutr. 2003; 77: 63-70.
- Levin BL, Varga E. MTHFR: Addressing Genetic Counseling Dilemmas Using Evidence-Based Literature. J Genet Couns. 2016; 25: 901-911.
- Leng S, Zhao A, Zhang J, et al. Methylenetetrahydrofolate Reductase Gene C677T Polymorphism-Dietary Pattern Interaction on Hyperhomocysteinemia in a Chinese Population: A Cross-Sectional Study. Front Cardiovasc Med. 2021; 8: 638322.
- Vijayan M, Chinniah R, Ravi PM, et al. MTHFR (C677T) CT genotype and CT-apoE3/3 genotypic combination predisposes the risk of ischemic stroke. Gene. 2016; 591: 465-470.
- Kang SS, Rosenson RS. Analytic Approaches for the Treatment of Hyperhomocysteinemia and Its Impact on Vascular Disease. Cardiovasc Drugs Ther. 2018; 32: 233-240.
- Liu X, Li Y, Wang M, et al. The 1316T>C missenses mutation in MTHFR contributes to MTHFR deficiency by targeting MTHFR to proteasome degradation. Aging (Albany NY). 2020; 13: 1176-1185.
- Iida S, Nakamura M, Asayama S, et al. Rapidly progressive psychotic symptoms triggered by infection in a patient with methylenetetrahydrofolate reductase deficiency: A case report. BMC Neurol. 2017; 17: 47.
- Diekman EF, de Koning TJ, Verhoeven Duif NM, Rovers MM, van Hasselt PM, et al. Survival and psychomotor development with early betaine treatment in patients with severe methylenetetrahydrofolate reductase deficiency. JAMA Neurol. 2014; 71: 188–194.