
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
Generalized edema as presenting feature of anti-NXP2 positive Juvenile dermatomyositis: A case report and review of literature
Mahdi Adouania1*; Thouraya Ben Younes1,2; Zouhour Miladi1; Hedia Klaa1,2; Hanene Benrhouma1,2; Aida Rouissi1; Ichraf Kraoua1,2; Malika Ben Ahmed2,3; Ilhem Ben Youssef-Turki1,2
1LR 18SP04 and Department of Child Neurology, National Institute of Neurology Mongi-Ben Hamida of Tunis, Tunisia.
2Faculty of Medicine of Tunis, University of Tunis El Manar. Tunisia.
3Department of Clinical Immunology, Pasteur Institute, Tunis, Tunisia.
*Corresponding Author : Mahdi Adouania
LR 18SP04 and Department of Child Neurology, National Institute of Neurology Mongi-Ben Hamida of Tunis, Tunisia.
Email: mahdi.adouania@gmail.com
Received : Aug 27, 2022
Accepted : Sep 19, 2022
Published : Sep 26, 2022
Archived : www.jcimcr.org
Copyright : © Adouania M (2022).
Abstract
Juvenile Dermatomyositis (JDM) is a rare childhood inflammatory disease. Clinical manifestations include muscle weakness and skin rashes. However, generalized edema is uncommon in JDM. Treatment is based on prolonged immunosuppressive therapy. Only few case reports of edematous JDM has been described. Here we aim to describe a case of edematous JDM with review of literature.
Citation: Adouania M, Younes TB, Miladi Z, Klaa H, Benrhouma H, et al. Generalized edema as presenting feature of anti-NXP2 positive Juvenile dermatomyositis: A case report and review of literature. J Clin Images Med Case Rep. 2022; 3(9): 2073.
Introduction
Juvenile Dermatomyositis (JDM) is a rare childhood systemic inflammatory disease affecting mainly the skin and the muscles [1]. Initial presentations of JDM can be heterogenous with symptoms including proximal muscle weakness, classic skin rashes such as Gottron’s papules or heliotrope rash in addition to localized periorbital edema [9]. However, generalized edema is a very uncommon clinical phenotype of JDM and has been described in rare case reports of children with JDM [7]. The recent immunological advance has contributed to a better understanding of dermatomyositis spectrum based on myositis specific antibodies [6]. Anti-NXP-2 autoantibody is the second most common antibody in JDM. Its clinical features include subcutaneous edema, dysphagia, calcinosis and a severe muscle phenotype characterized by myalgia and muscle weakness [6].
Here we are presenting a case of edematous juvenile dermatomyositis in a young 3 years old boy of Tunisian origin with a review of literature.
Case report
A 3-year-old boy was referred to our department for gait disturbance with frequent falls. He was born to non-consanguineous parents. His family history was unremarkable, and he had normal psychomotor milestones. He had a personal history of elbow skin abscess after a traumatic fall, treated with antibiotics with good outcome. He presented with progressive limb weakness and gait disturbance 2 months ago associated with mild facial edema. Ten days after the onset, appearance of generalized edema essentially in the face and upper limbs associated with dysphagia and myalgia.
Neurological examination showed waddling walk, symmetric proximal and distal muscle weakness, and weak deep tendon reflexes in the lower limbs.
Examination of the skin revealed the presence of confluent violaceous, edematous macules around eyelids, forehead, cheek (heliotrope rash) associated with generalized swelling predominantly in the face and neck, erythematous macules on the dorsum of the metacarpophalangeal joints (Gottron papules), Flagellate erythema on the back and alopecic plates (Figure 1).
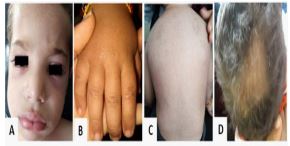
Routine laboratory tests including liver function tests, renal function tests, plasma glucose level, and serum electrolytes were normal. Lactate Dehydrogenase (LDH) level was elevated at 449 U/L (Normal value: < 250 U/L), Creatine Kinase (CK) level was also elevated at 256 U/L (Normal value: < 150 U/L). The search for proteinuria was negative. Abdominal ultrasound showed mild intraperitoneal effusion. Transthoracic echocardiography was normal. The diagnosis of Juvenile Dermatomyositis (JDM) was suspected. We completed by an electroneuromyogram which revealed a myopathic pattern. The search of Myositis-Specific Antibodies revealed an important positivity of antibodies anti- nuclear matrix protein NXP2 (+++) and low positivity of anti-Ku antibodies. Antinuclear ANTIBODY (ANA) was also positive.
Standard radiographs did not show calcinosis.
Muscular biopsy showed an in equal size of the fibers with the presence of an inflammatory parenchymal and perivascular infiltrate with increasing of the size of the connective tissue which dissociates the fibers. Some inflammatory infiltats obstruct the vascular wall.
Taking into consideration the clinical, electrophysiological, immunological, and histopathological findings, our patient was diagnosed with Anti-NXP2 positive Juvenile dermatomyositis.
Our Patient was treated with intravenous methylprednisolone (30 mg/kg/day) during 5 days followed by oral prednisolone (1.5 mg/kg/day) for 6 weeks then progressive tapering. Given the severity of the disease and lack of improvement after methylprednisolone, we indicated intravenous immunoglobulin pulse (0.4 g/kg/day) during five days repeated at one month. Our patient was also treated with Methotrexate at the dose of 15 mg/m2/week given by the subcutaneous route with folic acid supplementation.
Clinical follow-up showed a remission of the cutaneous manmanifestations, an improvement of muscle power and a normalization of muscle enzymes levels after 12 months of immunosuppressive treatment.
Table 1: Cases of JDM with generalized edema as clinical presentation.
| References | Case | Age (years) | Sex | Disease evolution | Severe muscle weakness | Myalgia | Dysphagia | Dysphonia | Skin or digestive ulcerations | Calcinosis | Antibodies | Response to treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [14] | 1 | 3.6 | M | 5 months | + | + | + | + | + | - | NA | - |
| [15] * | 2 | 17 | M | NA | + | + | + | + | + | - | NA | NA |
| [16]‡ | [3-11]‡ | Median :7.5 (2.3-10.6) |
5M 4F |
NA | (+) in 9/9 Severe weakness | 8/9 | 5/9 | NA | (+) in 1/9 NAin 8/9 |
4/9 | NA | 1/4 + 5/8 NA |
| [17] | [12-13] | Median : 2.2 | 1M 1F | 5 months | 2/2 : (+) ,2/2 : BB | 1 (+), 1 NA | NA | NA | 0/2 | 2/2 | NA | NA |
| [18] | 14 | 3.5 | F | NA | + , BB | + | NA | NA | - | - | NA | NA |
| [19] | 15 | 6 | F | NA | +, BB | + | NA | NA | + | - | NA | NA |
| [20] | 16 | 10 | F | NA | + , BB | + | + | NA | + | + | NA | NA |
| [21] | 17 | 7.1 | F | NA | + , BB | + | + | + | + | NA | NA | NA |
| [22] | 18 | 7 | F | 2 months | + , BB/WC | + | + | + | + | - | NA | - |
| [23] | 19 | 14 | M | 1 week | + | + | - | NA | - | - | NA | + |
| [24] | 20 | 8 | F | NA | + | + | + | NA | + | - | NA | - / death |
| [25] | 21 | 3.5 | M | 1 month | + , BB | + | + | + | + | - | NA | - |
| [26] ** | 22 | 6 | M | NA | - | + | - | - | + | - | NA | + |
| [27] | 23 | 16 | M | 1 month | + | + | + | - | + | NA | NA | + |
| [28] | 24 | 3.5 | M | 6 months | + | + | + | - | + | - | NA | + |
| [29] | 25 | 4 | M | 6 months | + | + | + | - | + | - | NA | + |
| [30] | 26 | 13 | F | 15 days | + | - | - | - | - | - | NA | + |
| [31] | 27 | 8 | M | 15 days | + | - | - | + | + | - | NA | + |
| [32] | 28 | 8 | M | 7 days | + | - | - | - | + | - | Anti-Jo1 | + |
| [33] | 29 | 4 | M | 6 weeks | + | + | + | + | + | - | NA | + |
| [34] | 30 | 7 | M | 2 weeks | + | + | - | - | - | - | NA | NA |
| [35] | [31-32] | 11 /4 | F/F | 2 weeks/ 4 months | +/+ | +/+ | -/- | +/- | +/+ | -/- | - / Anti-Ro60 | + / + |
| Our case | 33 | 3 | M | 4 months | + | - | + | - | + | - | Anti-NXP2 | + |
| Total | 33 | [2,3-17] | 19 M /14 F | 32/33 | 27/32 | 17/31 | 8/18 | 20/26 | 7/31 | 3/4 | 14/20 |
(+): Present; (-): Absent; NA: Non Available; BB: Bedbound; WC: wheel chair; M: male; F: female; ‡ In this series, 9 of 26 cases had generalized edema associated; *: This case of JDM was associated with mediastinal tumor; **: This case of JDM was associated with hereditary angioneurotic edema.
Discussion
This case illustrates a severe anti-NXP2 positive JDM with generalized oedema in a child. Juvenile dermatomyositis is a rare inflammatory autoimmune disease that belong to a very large heterogenous group of muscle diseases known as ‘Idiopathic Inflammatory Myopathies (‘IMM’s) [1]. Skin and muscle manifestations are often typical, however, generalized edema as clinical feature of JDM has been rarely reported. To the best of our knowledge, only 32 pediatric cases of JDM with anasarca has been reported in the English literature (Table 1) [14-35].
Taking into consideration the new IMM’s classification of the ACR EULAR 2017 [2], the criteria for JDM diagnosis in our patient were met. Myositis specific antibodies might be helpful as a diagnosis tool, however, only two cases in the literature of JDM patients presenting with anasarca were reported to have a positive myositis antibody [3,35].
At the time of diagnosis, our patient had some atypical features with generalized edema associated with mild abdominal as cites. Therefore, early recognition of anasarca is crucial and ruling out the differential diagnosis of edema in pediatric patients is pivotal before considering JDM diagnosis [4]. In our patient, cardiac, renal, and hepatic causes of anasarca were ruled out.
The pathogenesis remains unclear, although it has been suggested that intense inflammatory activity with activation of complement may lead to vascular disease with muscle microinfarcts and as a result increase capillary permeability [5].
Muscle involvement and dysphagia are frequently associated with edematous juvenile dermatomyositis as observed in the case we report.
This severe form of generalized edema is not clearly correlated with autoantibody status in juvenile form of Dermatomyositis (DM), although subcutaneous edema is more common in adult patients with anti-NXP2 positive DM than pediatric patients [6].
Myositis associated antibodies are not well described in pediatric DM. However, antinuclear antibody positivity, as seen in our patient, can be found up to 68% of JDM cases and is considered as risk factor for disease complications such as growth retardation and lipodystrophy [7]. As for anti-Ku antibody positivity, it has been linked to overlap syndroms without a specific JDM feature [8]. Our patient had a low positivity of anti-Ku antibodies.
Anti-NXP2 positive JDM patients have a specific clinical spectrum with 43% of them developing calcinosis within 2 years after first symptoms onset requiring an early control of the disease [9].
Calcinosis is one of the main disease complications in anti-NXP2 JDM patients alongside with increased risk of gastrointestinal bleeding, ulcers and dysphagia. In our case, no calcinosis was detected based on bones standard radiographs after 10 months of symptoms onset.
(+): Present; (-): Absent; NA: Non Available; BB: Bedbound; WC: wheel chair; M: male; F: female; ‡ In this series, 9 of 26 cases had generalized edema associated; *: This case of JDM was associated with mediastinal tumor; **: This case of JDM was associated with hereditary angioneurotic edema.
There is no conventional treatment for edematous JDM, however, as it is the case for our patient, the standard approach is to use early and prolonged immunosuppressive treatment based on addition of methotrexate, together with an aggressive taper of corticosteroid therapy alongside with IVIG pulses in refractory forms [11]. There is no high-level evidence of when to stop therapy, however, if a patient has been off steroids and in remission on methotrexate for a minimum of 1 year, with drawing treatment could be considered [12]. Our patient haven’t been yet off steroids and is still receiving weekly injections of MTX.
Conclusion
Edematous form of JDM is very rare and uncommon as inaugural clinical presentation. The presence of NXP-2 antibodies is known to be associated with a severe phenotype including generalized edema. An early diagnosis with an aggressive and prolonged therapeutic approach is required to avoid disease related morbidity and improve functional prognosis.
Declarations
Statement of authors’ contribution:
• All authors contributed equally to the study conception and\or study design and\or data collection and\or data analysas and\or results interpretations and\or manuscript writing and\or manuscript correction and\or proof Reading and\ or manuscript submission.
• All authors have agreed to this final version of the paper being submitted to the European Journal of Paediatric Neurology.
Conflict of interest
• Declaration of conflicting interests: The Author(s) declare(s) that there is no conflict of interest.
• Funding acknowledgement : The author(s) received no financial support for the research, authorship, and/or publication of this article.
• Funding acknowledgement : The author(s) received no financial support for the research, authorship, and/or publication of this article.
• Financial benefits to the authors: none.
Ethical considerations
• Free and informed consent for participation and publication of the parents accompanying the child with CP was obtained in writing after using forms detailing the purpose and modalities of participation.
• Confidentiality was respected by an anonymous coding of the files that were to be kept anonymously in a specific binder.
• We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- Mariam Pillai K, et al. Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol. 2018; 75: 1528-1537.
- Lundberg IE et al. International Myositis Classification Criteria Project consortium, The Euromyositis register and The Juvenile Dermatomyositis Cohort Biomarker Study and Repository (JDRG) (UK and Ireland). 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017; 76: 1955-1964.
- Shelley BP, Chakraborty S. A viral polymyositis masquerade: Life-Threatning case of juvenile dermatomyositis complicated by systemic capillary leak syndrome. Ann Indian Acad Neurol. 2018; 21: 70–74.
- Leung AK, Robson WL. Oedema in childhood. J R Soc Promot Health. 2000; 120: 212-219.
- R. Goussot, C. Wettlé, C. Le Coz, B. Cribier, D. Lipsker. Severe edematous dermatomyositis. 2016.
- Albayda J, Pinal Fernandez I, Huang W, Parks C, Paik J, et al. Antinuclear Matrix Protein 2 Autoantibodies and Edema, Muscle Disease, and Malignancy Risk in Dermatomyositis Patients. Arthritis Care & Research. 2017; 69: 1771–1776.
- Barut K, Aydin PO, Adrovic A, Sahin S, Kasapcopur O, et al. Juvenile dermatomyositis: Atertiary center experience. Clin Rheumatol. 2017.
- Cotter C, Rudd E, Williamson E, Philippidou M, Tewari A. Anti-Ku-positive juvenile dermatomyositis. Clin Exp Dermatol. 2021.
- Wu JQ, Lu MP, Reed AM. Juvenile dermatomyositis: Advances in clinical presentation, myositis-specific antibodies and treatment. World J Pediatr. 2020;16: 31-43.
- Tu J, McLean-Tooke A, Junckerstorff R. Increasing recognition of dermatomyositis with subcutaneous edema - isthis a poorer prognostic marker. Dermatol Online J. 2014; 20: 21244.
- Wu Q, Wedderburn LR, Mc Cann LJ. Juvenile Dermatomyositis: Latest Advances. Best Pract Res Clin Rheumatol. 2017; 31: 535–557
- Rosa Neto NS, Goldenstein-Schainberg C. Juvenile dermatomyositis: review and update of the pathogenesis and treatment. Rev Bras Reumatol. 2010; 50: 299-312.
- Giancane G, et al. Paediatric Rheumatology International Trials Organization (PRINTO). The PRINTO evidence-based proposal for glucocorticoids tapering/discontinuation in new onset juvenile dermatomyositis patients. Pediatr Rheumatol Online J. 2019; 17: 24.
- Cook CD, Rosen FS, Banker BQ. Dermatomyositis and focal scleroderma. Pediatr Clin North Am. 1963; 10: 979–1016.
- Sheldon JH, Young F, Dyke SC. Acute dermatomyositis associated with reticulo-endotheliosis. Lancet. 1939; 1: 82–85
- Wedgewood RJ, Cook CD, Cohen J. Dermatomyositis: report of 26 cases in children with a discussion of endocrine therapy in 13. Pediatrics. 1953; 12: 447–466.s
- Hecht MS. Dermatomyositis in childhood. J Pediatr. 1940; 17: 791–800.
- Steiner WR. Dermatomyositis, with report of two cases. JAMA. 1922; 78: 271–273.
- Rothstein JL, Welt SK. Calcinosis universalis and calcinosis circumscripta in infancy and in childhood. Three cases of calcinosis universalis, with a review of the literature. Am J Dis Child. 1936; 52: 368–422.
- Karelitz S, Welt SK. Dermatomyositis. Am J Dis Child. 1932; 43: 1134–1149.
- Selander P. Dermatomyositis in early childhood. Acta Med Scand Suppl. 1950; 246: 187–203.
- Mitchell JP, et al. Juvenile dermatomyositis presenting with anasarca: A possible indicator of severe disease activity. J Pediatr. 2001; 138: 942–945.
- Karabiber H, Aslan M, Alkan A, Yakinci C. A rare complication of generalized edema in juvenile dermatomyositis: a report of one case. Brain Dev. 2004; 26: 269–272.
- Mehndiratta S, Banerjee P. Juvenile dermatomyositis presenting with anasarca. IndianPediatr. 2004; 41: 752–753.
- Zedan M, el-Ayouty M, Abdel-Hady H, Shouman B, el-Assmy M, Fouda A, et al. Anasarca: Not a nephrotic syndrome but dermatomyositis. Eur J Pediatr. 2008; 167: 831–834.
- Narasimhan R, Lakshman R, Amos RS, Williams LH, Egner W, Finn A, et al. Juvenile dermatomyositis associated with hereditary angioneurotic oedema. Arch Dis Child. 2002; 87: 563.
- Carlos EP LúcioFilho ,Juvenile dermatomyositis presenting with anasarca: A possible new variant. Acta Reumatologica Portuguesa. 2008; 33: 238-242.
- Wakhlu A, et al. Lack of neck holding and anasarca: Important but ignored manifestations of juvenile dermatomyositis. Indian. J Rheumatol. 2013; 8: 139– 141.
- Saygi S, Alehan F, Baskin E, Bayrakci US, Ulu EM, Ozbek N, et al. Juvenile dermatomyositis presenting with anasarca. J Child Neurol. 2008; 23: 1353- 1356.
- Torres Jiménez AR, Solís Vallejo E, Céspedes Cruz AI, Sánchez Uribe M, et al. Subcutaneous edema in juvenile dermatomyositis. Reumatol Clin (Engl Ed). 2019; 15: e49-e50.
- Sharma D, Singh S, Suri D, Rawat A, Shava U, Sodhi KS, et al. Anasarca as the initial presentation of juvenile polymyositis: an uncommon occurrence. Rheumatol Int. 2012; 32: 2589-2590.
- Shelley BP, Chakraborti S. A Viral Polymyositis Masquerade: Life-Threatening Case of Juvenile Dermatomyositis Complicated by Systemic Capillary Leak Syndrome. Ann Indian Acad Neurol. 2018; 21: 70-74.
- Chandrakasan S, Singh S, Ratho RK, Kumar S, Mishra B. Anasarca as the presenting manifestation of parvovirus B19 associated juvenile dermatomyositis. Rheumatol Int. 2009; 29: 565-567.
- Nickavar A, Mehr AM. Nephrotic syndrome and juvenile dermatomyositis. Rheumatol Int. 2012; 32: 2933–2935.
- Schildt EE, De Ranieri D. Anasarca as the presenting symptom of juvenile dermatomyositis: A case series. Pediatr Rheumatol. 2021; 19: 120.