
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
TSC1 mutation in neonatal tuberous sclerosis: A case report
Jiwon Moon1; Hong Yeon Lee1; Jisook Yim2; Myungshin Kim2; In Yang Park3; Ji Hye Jo1*
11Department of Obstetrics and Gynecology, Yeouido St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea.
2Department of Laboratory Medicine, College of Medicine, The Catholic University of Korea, Republic of Korea.
2Catholic Genetic Laboratory Center, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea.
3Department of Obsterics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea.
*Corresponding Author : Ji Hye Jo, MD
Department of Obstetrics and Gynecology, Yeouido St. Mary’s Hospital, College of Medicine, The Catholic University of Korea Republic of Korea.
Tel: +82-3-779-1613, Fax: +82-3-779-1614;
Email: jjjihhh.jo@gmail.com
Received : Oct 17, 2022
Accepted : Nov 01, 2022
Published : Nov 08, 2022
Archived : www.jcimcr.org
Copyright : © Ji Hye Jo (2022).
Abstract
A 32-year old woman, gravida 1 para 0, was referred to Seoul St. Mary’s Hospital for fetal cardiac mass by ultrasound at 38 weeks of gestation from a local obstetric office. Detailed fetal echocardiogram was performed and the presence of multiple cardiac tumors was observed, suggesting rhabdomyoma. After birth, fetal echocardiography and genetic screening were performed. A targeted Sanger sequencing analysis was performed for TSC1 and TSC2 genes. Molecular analyses revealed a heterozygous gene variant with a novel frame shift mutation in TSC1, confirming the diagnosis of Tuberous Sclerosis (TSC). A segregation study of the causal mutation was performed by Sanger sequencing for both parents, revealing no mutation in either parent. Thus, the novel frameshift mutation occurred de novo. For fetuses with suspected cardiac rhabdomyoma identified by ultrasound, perinatal genetic testing for TSC should be performed for both the fetus and family members for early detection, early diagnosis, and better prognosis. Early diagnosis of TSC can provide greater opportunities for infants to obtain timely neonatal treatment and better outcome.
Keywords: Tuberous sclerosis; Rhabdomyoma; Fetal ultrasonography; TSC1 protein.
Citation: Moon J, Lee HY, Yim J, Kim M, Park IY, Jo JH, et al. TSC1 Mutation in Neonatal Tuberous Sclerosis : A Case report. J Clin Images Med Case Rep. 2022; 3(11): 2146.
Introduction
Tuberous Sclerosis Complex (TSC) is an autosomal dominant multisystemic neurocutaneous disorder characterized by noncancerous tumors in various organ systems of the body [1]. The brain, skin, heart, kidney, lungs, and eyes are frequently affected. Although classic manifestations of TSC include mental retardation, epilepsy, and sebaceous adenoma, signs, symptoms, and severity of this disorder vary from one patient to another due to specific organs involved. The incidence of TSC has been estimated to be 1 in 6,000 to 10,000 live births [2].
TSC is caused by mutations of tumor suppressor gene TSC1 or TSC2. TSC1 gene is located on chromosome 9q34. It encodes a growth inhibitory protein named hamartin. TSC2 is located on chromosome 16p13.3. It provides instructions for producing tuberin [3,4]. Hamartin and tuberin are known to suppress cell growth by inhibiting the activation of mTOR (mammalian target of rapamycin), a serine/threonine protein kinase that reconfigures cellular metabolism and regulates translation, cytokine responses, antigen presentation, macrophage polarization and cell migration [5,6]. In TSC patients, mTOR is dysregulated due to changes of hamartin or tuberin, leading to abnormal differentiation and development of cells. In this report, we present a newborn with multiple cardiac rhabdomyoma who showed a de novo mutation in TSC1 gene.
Case presentation
A 32-year old woman, gravida 1 para 0, was referred to Seoul St. Mary’s Hospital for a fetal cardiac mass by ultrasound at 38 weeks of gestation from a local obstetric office.The mother had no history of drug ingestion, alcohol use, or smoking during pregnancy. There was no familial history of genetic or neurodevelopmental disorders. There was no other antenatal problem.
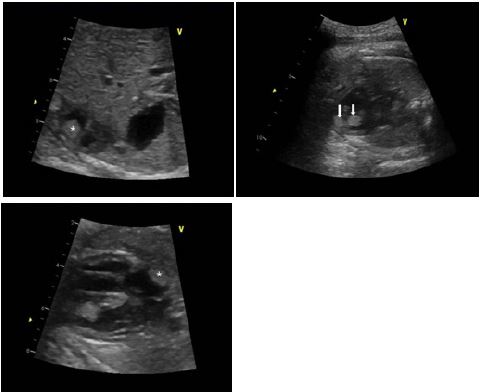
Detailed fetal echocardiogram was performed and multiple cardiac tumors (one in right atrium, two in right ventricle, and two in left ventricle, Figure 1) were observed, suggesting rhabdomyoma. Ventricular function seemed to be normal. Our multidisciplinary team planned to perform cardiac ultrasound after birth.
She delivered a female infant at 39 weeks and 3 days of gestation through an emergency cesarean section owing to non-reassuring fetal status. The infant weighted 3590 g. Her 1-minute and 5-minute Apgar scores were 6 and 9, respectively. After birth, the infant was admitted to the neonatal intensive care unit for thorough examination. One day after birth, fetal echocardiography and genetic screening were performed.
Cardiac ultrasound
Cardiac ultrasound revealed the presence of at least five cardiac tumors in the fetus. One elliptical mass was localized in the right atrium. The mass did not interfere with Superior Vena Cava (SVC) or Inferior Vena Cava (IVC) flow. Two masses with sizes of 0.7 cm and 0.6 cm were found in the apex of right ventricle. Two masses of 0.8 cm and 0.4 cm were detected in the left ventricle. These masses were attached to the septum and connected to each other. These observations strongly suggested multiple cardiac rhabdomyomas. There was no obstruction of Left Ventricular Outflow Tract (LVOT) or Right Ventricular Outflow Tract (RVOT). The ventricular function was observed to be normal.
Genetic screening
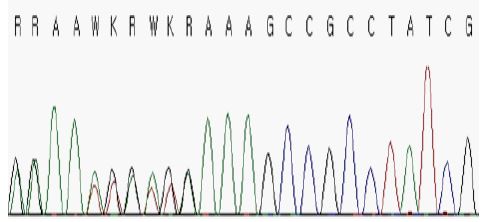
Definite features of TSC strongly suggested the presence of a genetic modification in one of the two genes, TSC1 and TSC2, commonly associated with the disease. A targeted Sanger sequencing analysis was performed for TSC1 and TSC2 genes. Informed consent was obtained from the patient’s parents before genetic analyses were conducted. Resutls of molecular analyses revealed a heterozygous c.2643dup variant (NM_000368.4) that led to a p.(Ala882Serfs*22) novel frameshift mutation in TSC1, thus confirming the diagnosis of TSC (Figure 2). A segregation study of the causal mutation was performed by Sanger sequencing for both parents, revealing no mutation in either parent (Figure 2). Thus, the mutation had a de novo origin.
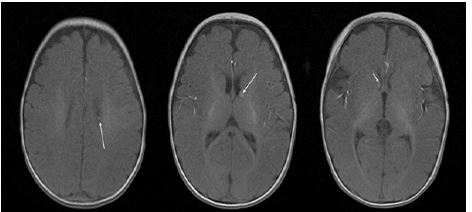
After detecting TSC1 mutation, detailed examination was performed to find other manifestations associated with TSC. Her brain MRI showed a few small, T1W high SI nodular lesions in both subependymal zones, suggesting tuberous sclerosis involvement and subependymal hamartomas (Figure 3). Whole body spine and extremities X-ray revealed no significant bony abnormality. Other examinations including Visual Evoked Potentials (VEP)/Auditory Evoked Potentials (AEP) and kidney sonography were also normal. The infant is followed up right now on an outpatient basis.
Discussion
TSC is a multisystem disorder with a highly variable phenotype characterized by hamartomas in multiple organ systems including the brain, heart, skin, lungs, and kidneys. TSC is known to be caused by autosomal dominant mutation of TSC1 located on chromosome 9q34 or TSC2 gene located on chromosome 16p13.3. TSC1 and TSC2 gene mutations account for 31% and 67% of TSC mutations, respectively. These variants often occur de novo, attributing 70-80% of all TSC.
The 2012 International Tuberous Sclerosis Complex Consensus Recommendations have added genetic diagnosis to the diagnostic criteria for TSC and pointed out that genetic diagnosis can be used as an independent diagnostic criteria [7]. Primary cardiac tumor is rare with a prevalence of 0.25% in infants and young children. Cardiac rhabdomyoma is the most common cardiac tumor, accounting for 60% of such cases. Fetal cardiac rhabdomyoma is usually detected after 20 weeks of gestation. It frequently occurs in the ventricular cavity. It is well known that cardiac rhabdomyoma and TSC are strongly related regardless of the number of tumors. With the development of prenatal ultrasound technology, an increasing number of fetal cardiac tumors have been reported in recent years [8,9].
In our case, the fetus showed rhabdomyoma prenatal features that continuously existed after birth. We immediately performed a targeted Sanger sequencing analysis to rule out TSC. Early detection of de novo mutation can lead to monitoring of TSC for the infant. The patient is followed up currently without other developmental disabilities.
Conclusion
In conclusion, we present this case to alert the importance of antenatal diagnosis of TSC by fetal cardiac rhabdomyomas. For fetuses with suspected cardiac rhabdomyoma identified by ultrasound, perinatal genetic testing for TSC is recommended for both fetus and family members for early detection, early diagnosis, and better prognosis. Early diagnosis of TSC can provide greater opportunities for infants to obtain early neonatal treatment and better outcome.
References
- Rosset C, Netto CBO, Ashton Prolla P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: A review. Genet Mol Biol. 2017; 40: 69-79.
- Wataya Kaneda M, Uemura M, Fujita K, Hirata H, Osuga K, Kagitani Shimono K, et al. Tuberous sclerosis complex: Recent advances in manifestations and therapy. Int J Urol. 2017; 24: 681-691.
- Avgeris S, Fostira F, Vagena A, Ninios Y, Delimitsou A, Vodicka R, et al. Mutational analysis of TSC1 and TSC2 genes in Tuberous Sclerosis Complex patients from Greece. Sci Rep. 2017; 7: 16697.
- Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M, et al. Genetic Etiologies, Diagnosis, and Treatment of Tuberous Sclerosis Complex. Annu Rev Genomics Hum Genet. 2019; 20: 217-240.
- Sampson JR. TSC1 and TSC2: Genes that are mutated in the human genetic disorder tuberous sclerosis. Biochem Soc Trans. 2003; 31: 592-596.
- Sadowski K, Kotulska K, Schwartz RA, Jóźwiak S. Systemic effects of treatment with mTOR inhibitors in tuberous sclerosis complex: A comprehensive review. J Eur Acad Dermatol Venereol. 2016; 30: 586-594.
- Portocarrero LKL, Quental KN, Samorano LP, Oliveira ZNP, Rivitti-Machado M. Tuberous sclerosis complex: Review based on new diagnostic criteria. An Bras Dermatol. 2018; 93: 323-331.
- Chen L, Jiang Y, Wang J. Fetal cardiac rhabdomyoma due to paternal mosaicism of TSC2: A case report. Medicine (Baltimore). 2020; 99: e21949.
- Pruksanusak N, Suntharasaj T, Suwanrath C, Phukaoloun M, Kanjanapradit K, et al. Fetal cardiac rhabdomyoma with hydrops fetalis: Report of 2 cases and literature review. J Ultrasound Med. 2012; 31: 1821-1824.