
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
CNV and RNA analysis reveal a germline pathogenic duplication of MSH2 exon 15 in a family with Lynch syndrome: A case report
Despina Apostolopoulou1; Mustafa Özdoğan2; Konstantinos Agiannitopoulos1*; Georgia Pepe1; Kevisa Potska1; Georgios N Tsaousis1; Nikolaos Tsoulos1; Eleni Patsea3; Onder Kırca2; Muharrem Okan Çakır4; Eirini Papadopoulou1; George Nasioulas1
1Genekor Medical S.A, 52 Spaton Ave, 15344, Athens, Greece.
2Division of Medical Oncology, Memorial Hospital,07025 Kepez, Antalya, Turkey.
3Department of Pathology, Metropolitan General Hospital, 15562,Cholargos, Greece.
4Cancer Theme, Department of Biomolecular Sciences, Kingston University London, KT1 2EE, Kingston upon Thames, UK.
*Corresponding Author : Konstantinos Agiannitopoulos
Genekor Medical SA, 52 Spaton Ave, 15344, Athens, Greecee.
Email: kagiannitopoulos@genekor.com
Received : Jan 23, 2023
Accepted : Feb 09, 2023
Published : Feb 16, 2023
Archived : www.jcimcr.org
Copyright : © Agiannitopoulos K (2023).
Abstract
Background: MSH2 germ line pathogenic variants are a well-recognised cause of Lynch syndrome, predisposing individuals to a variety of malignancies, most usually colorectal and endometrial cancer. Partial duplications of MSH2 gene, due to their position in the genome and frequently unclear mechanisms of pathogenicity, are often classified as Variants of Uncertain Significance (VUS).
Case presentation: CNV (Copy Number Variation) analysis revealed a duplication of MSH2 exon 15 in a young male patient with colorectal cancer as well as in his affected family members. RNA analysis elucidated the impact of this duplication on RNA, revealing that it leads to an abnormal transcript, thus providing experimental evidence for a pathogenic effect.
Conclusions: We show that the combination of CNV and RNA analysis provides critical information for the identification and proper classification of pathogenic/ likely pathogenic variants which, in turn, is of great importance for the patients as well as for their family members with an actionable impact in clinical practice.
Keywords: Colon cancer; RNA study; Copy number variation; MSH2 duplication; Variant classification.
Abbreviations: VUS: Variants OF Uncertain Significance; CNV: Copy Number Variation; MMR: Mismatch Repair; MSI: Microsatellite Instability; CMMRDS: Constitutional Mismatch Repair Deficiency Syndrome; FFPE: Formalin-Fixed and Paraffin-Embedded; IHC: Immunohistochemistry; NGS: Next Generation Sequencing.
Citation: Apostolopoulou D, Özdoğan M, Agiannitopoulos K, Pepe G, Potska K, et al. CNV and RNA analysis reveal a germline pathogenic duplication of MSH2 exon 15 in a family with Lynch syndrome: A case report. J Clin Images Med Case Rep. 2023; 4(2): 2289.
Background
MSH2 belongs to the Mismatch Repair (MMR) proteins, which are responsible for the repair of DNA replication errors. These errors preferentially accumulate in regions of repetitive DNA sequences called microsatellites, causing Micro Satellite Instability (MSI) [1]. MSI is a main characteristic of Lynch Syndrome (LS), one of the most common hereditary cancer syndromes which are inherited in an autosomal dominant manner. LS, previously known as Hereditary Nonpolyposis Colorectal Cancer (HNPCC), is caused by germline mutations of the MMR genes MLH1, MSH2, MSH6, and PMS2 and, rarely, deletions of the 3’ UTR region of the non-MMR gene EPCAM, which lead to hypermethylation of the MSH2 promoter and loss of MSH2 expression [2]. Biallelic inherited MMR pathogenic variants in MLH1, MSH2, MSH6, and PMS2 are associated with a rare condition called constitutional mismatch repair deficiency syndrome (CMMRDS) [3].
Patients with LS have up to an 80% lifetime risk of developing colon cancer and, in women, a 60% lifetime risk of developing endometrial carcinoma [4]. Apart from colorectal and endometrial cancers, LS related cancers include gastric, ovarian, pancreas, urothelial (kidney, renal pelvis, ureter, bladder and prostate), brain, biliary tract, small intestinal cancers, as well as sebaceous adenomas, sebaceous carcinomas and keratoacanthomas [5].
Approximately 40% of the pathogenic germline variants causative for LS are in MSH2 gene [4]. Structural variants such as inversions and Copy Number Variations (CNVs) are more common in MSH2 gene than in other MMR genes [6]. Although inversions and deletions are, in majority, classified as pathogenic, exonic duplications are, due to their position in the genome and frequently unclear mechanisms of pathogenicity, often classified as Variants of Uncertain Significance (VUS) [6].
Here, we present a family with Lynch Syndrome and different types of cancer with a pathogenic duplication of MSH2 exon 15.
Case presentation
Patient
A 23-year-old male of Turkish origin diagnosed with colorectal cancer was referred to our private diagnostic laboratory for genetic testing with a hereditary cancer panel. Peripheral blood samples from the proband and his family members, when available, were drawn for diagnostic purposes after obtaining a signed informed consent and permission for the anonymous use of their data for research purposes and/or scientific publications.
Gene testing
Genomic DNA was extracted from peripheral blood using Mag Core® Genomic DNA Whole Blood Kit (RBC Bioscience) according to the manufacturer’s instructions. The analysis of genes involved in hereditary cancer predisposition was performed using a solution-based capture approach. Targeted Next Generation Sequencing (NGS) was performed with a panel of 36 genes (Roche Nimble Gen Seq Cap EZ Choice) consisting of: APC (NM_000038), ATM (NM_000051), BARD1 (NM_000465), BMPR1A (NM_004329), BRCA1 (NM_007294), BRCA2 (NM_000059), BRIP1 (NM_032043), CDH1 (NM_004360), CDK4 (NM_000075), CDKN2A (NM_000077), CHEK2 (NM_007194), EPCAM (NM_002354), FANCA (NM_000135), FANCM (NM_020937), HOXB13:c.251G>A p.(G84E) (NM_006361), MEN1 (NM_000244), MLH1 (NM_000249), MRE11 (NM_005591), MSH2 (NM_000251), MSH6 (NM_000179), MUTYH (NM_001128425), NBN (NM_002485), NF1 (NM_000267), PALB2 (NM_024675), PMS2 (NM_000535), POLD1 (Exons 8-13) (NM_001256849), POLE (Exons 1-14) (NM_006231), PTEN (NM_000314), RAD50 (NM_005732), RAD51C (NM_058216.2), RAD51D (NM_002878.3), RET (NM_020975), SMAD4 (NM_005359), STK11 (NM_000455), TP53 (NM_000546) and VHL (NM_000551) [7]. The sample preparation was performed according to the manufacturer’s instructions in the Seq Cap EZ Choice Library User’s Guide (Roche Nimble Gen, Pleasanton, CA, USA).Sequencing was carried out using the Mis eq Illumina NGS (Illumina, San Diego, CA, USA) technology and sequence changes were identified and interpreted in the context of a single clinically relevant transcript using the commercially available software suite Seq Nextversion 4.4.0 (JSI Medical Systems GmbH, Ettenheim, Germany). The presence of CNVs was investigated using the commercial computational algorithm Seq Pilot (JSI medical systems GmbH, Germany) and verified by the use of MLPA method (Multiplex Ligation-dependent Probe Amplification, MRC Holland). Sample preparation of the proband, his family members and normal references was performed according to the manufacturer’s instructions. Briefly, specific MLPA probes were hybridized to each denatured DNA sample, followed by ligation of the hybridized probes, PCR amplification of ligated probes and fragment separation by capillary electrophoresis. Results were analysed with Coffalyser. Net.
Immunohistochemistry MMR and MSI analysis
Immunohistochemical (IHC) examination and evaluation of Mismatch Repair proteins (MMR) in the paraffin-embedded tissue was performed using the following antibodies: clone Μ1, ROCHE (MLH1 Ab), clone G219-1129, ROCHE (MSH2 Ab), clone SP93, ROCHE (MSH6 Ab), clone A16-4, ROCHE (PMS2 Ab) according to methodology described else were [8,9].
For the MSI analysis, genomic DNA was isolated from proband’s FFPE tumor biopsies using the Mag MAX™ Total Nucleic Acid Isolation Kit (Thermo Fischer Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The nucleic acid isolation was conducted in the areas of the FFPE block with the majority of Tumor Cell Content (TCC), as indicated by experienced pathologists in hematoxylin and eosin stained sections. Minimum required TCC was over 20%, in a tumor area of > 4 mm2. Microsatellite analysis was conducted using the Ion Ampli Seq™ Microsatellite Instability Panel (Thermo Fischer Scientific) a NGS based assay analyzing 76 markers to assess Microsatellite Instability (MSI) status in tumor-only and tumor-normal samples as indicated by the manufacturer. Analysis of the sequencing output from this panel was carried out using the “MSICall” plugin in the Torrent Suite.
RNA analysis
In order to investigate the impact of this variant on RNA level, total RNA was extracted from peripheral blood lymphocytes using Trizol (Invitrogen, Paisley, UK) following a standard pro tocol. C DNA was synthesized using the Super Script™ VILO™ c DNA Synthesis Kit (Thermo Fisher Scientific) as described by the manufacturer. The resulting c DNA was amplified using specific primers designed on MSH2 cDNA (NM_000251.1) so that the forward primer was downstream of the reverse primer within the duplicated region (MSH2EX15dup Forward primer: 5’–GCT AAA CAG AAA GCC CTG GAA C–3’, MSH2EX15dup Reverse primer: 5’–TAG CAA GCT CTG CAA CAT GAA–3’). The PCR product was purified using the Nucleo Fast® 96 PCRCl eanupkit (Macherey-Nagel GmbH and Co., Düren, Germany). The purified PCR product was used for each sequencing reaction performed using the BigDye® Terminator v1.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequencing reaction products were purified prior to electrophoresis using the Montage™SEQ96 Sequencing Reaction kit (EMD Millipore Corp., Billerica, MA, USA) and sequenced using a Seq Studio Genetic Analyzer (Applied Biosystems)
Results
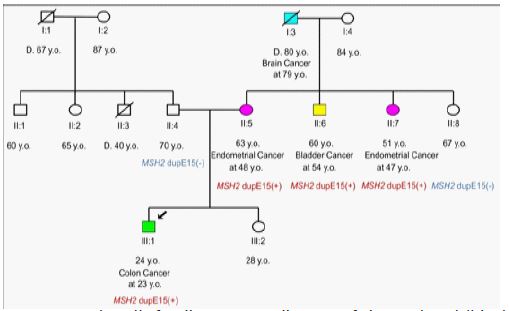
The proband, a 23-year-old male of Turkish origin diagnosed with colorectal cancer at the age of 23 had a strong family history of endometrial, bladder and brain cancer from his mother’s side (Figure 1).
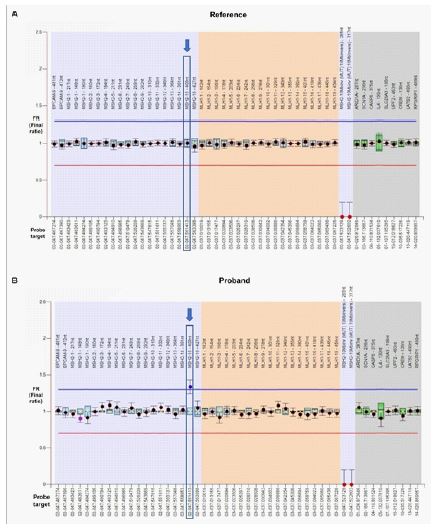
CNV analysis revealed that the proband carried a heterozygous duplication of exon 15 in MSH2 gene [NG_007110 (NM_000251): c.(2458+1_2459-1)_(2634+1_2635-1)dup], confirmed by MLPA microdeletion/microduplication analysis (MRC Holland, Amsterdam, Netherlands, SALSA KIT P003-D1) (Figure 2). No other clinically relevant variants were found. MLPA analysis of family members (affected and unaffected) showed that in this family the variant segregates with the MSH2-associated cancer [the patient’s mother (II: 5) and her two affected siblings (II: 6, II:7) carried the variant, whereas the unaffected father (II:4), sister (III:2) and maternal aunt (II:8) of the patient were normal for the above-mentioned variant) (Figure 1).
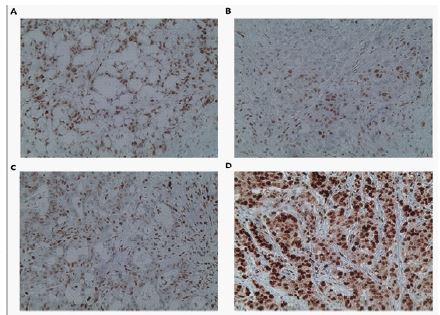
Immunohistochemical examination of MMR proteins in the proband’s Formalin-Fixed And Paraffin-Embedded (FFPE) tumor tissue indicated significantly reduced expression of the MSH2 protein (< 10%), while the expression of MLH1, MSH6, and PMS2 proteins was 60%, 70%, and 100%, respectively (Figure 3). During MSI analysis of the patient’s tumor tissue MSI-high was observed (MSI score=111,19). Α sample is considered positive if the MSI score is >30. The sample therefore was classified as Microsatellite-High (MSI-H).
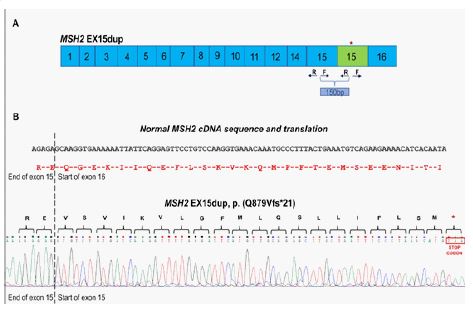
RNA analysis revealed that MSH2 exon 15 duplication was in tandem leading to a frame shift and a premature stop codon a few amino acid residues downsteam the end of exon 15 [MSH2 EX15dup, p.(Q879Vfs*21)] (Figure 4).
Discussion and conclusions
Duplication of MSH2 exon 15 has been described before in families affected with Lynch syndrome (HNPCC) [10,11] but, to our knowledge, no experimental studies were performed concerning this variant. In addition, the mutation database Clin Var [12] contains entries for different MSH2 multiple exons duplications which include exon 15 listed as variants of uncertain clinical significance (Variation IDs: 1002962, 831548, 1002963, 1027142, 1470345). Although MSH2 exon 15 duplication was found to segregate with cancer in the proband’s family, further evidence was needed to classify this variant as pathogenic.
CNV analysis alone, although very important to reveal the structural variant, could not elucidate the impact of this MSH2 partial duplication on functional level, so RNA analysis was crucial towards that direction. RNA analysis contributed to the classification of the MSH2 exon 15 duplication as pathogenic. It revealed that this variant leads to a frame shift and the creation of a novel translational termination codon 21 residues later, thus resulting in a truncated and non-functional protein product. Several variants resulting in premature stop codon in the neighbouring regions of MSH2 gene have been reported as pathogenic (Clin Var Variation IDs: 428483, 993962, 1392097). According to the ACMG/Clin Gen recommended guidelines for the classification of CNV variants [13], the MSH2 exon 15 duplication found in the proband and his affected family members was classified as pathogenic with a total score of 1.2 and the following evidence used: 1A, 2I (PVS1, assigned points: 0.9), 3A, 4F, 5D. RNA analysis, which proved that the duplication was in tandem leading to a disrupted reading frame, allowed the use of PVS1 for 2I, since according to the recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion, if a duplication of a portion of the gene of a defined length is inserted in tandem, one can predict if the reading frame will be disrupted leading to NMD, in which case PVS1 can be applied [14] (Clin Var submission: SUB12060189). RNA analysis in patients with Lynch Syndrome carrying different exon duplications in MSH2 gene provided evidence for the reclassification of these variants, originally classified as VUS, to pathogenic variants[15].
Immunohistochemistry (IHC) analysis of MMR proteins in the proband’s tumor tissue indicated significantly reduced expression of the MSH2 protein but the expression of MSH6 was retained. Although, in MSH2 deficient tumors, loss of expression of MSH6 protein is expected, intact or intermediate expression of MSH6 in IHC staining in tumors from Lynch syndrome patients with confirmed deleterious mutations in MSH2 has been observed [16,17]. It has been suggested that in tumors with intact MSH6, it is possible that the second MSH2 hit impaired MSH2 function but retained its ability to bind and stabilize MSH6 [16]. Indeed, NGS based MSI analysis performed in DNA from the proband’s tumor biopsies clearly showed MSI-high. This indicates the importance of NGS based MSI analysis, especially in cases like this, because it can clarify the presence of MSI-High when the IHC analysis for MMR proteins is ambiguous.
As in this case, the significance of proper classification of partial MSH2 gene duplications, by performing RNA analysis is of great importance and with a great impact in the management of the patient as well as for his family members, affecting the screening procedure, targeted therapies and decisions for risk-reducing interventions [18]. The proband, since he was a young-onset, node-positive stage III left colon cancer patient underwent left hemicolectomy: pT3N1 (2/38). Since MSI-high status did not predict unresponsiveness to chemotherapy in stage III colon cancer, the patient was administered 12 courses of adjuvant FOLFOX [19,20]. Adjuvant treatment was completed in six months and clinical follow-up was initiated.In addition, since MSI-high was observed during MSI analysis of the patient’s tumor tissue, the patient is eligible for immunotherapy administration [21].
According to the surveillance/prevention strategies of the NCCN guidelines for carriers of pathogenic/likely pathogenic variants colonoscopy every 2 years for colon cancer screening and Esophago Gastro Duodenoscopy (EGD) every 3-5 years for gastric cancer screening were recommended for the proband and his MSH2 positive family members, as well as annual gynaecological examination for female individuals [5]. In addition, it was recommended that they continue with other cancer screenings in accordance with their age. Last but not least, knowledge of the genetic background allows the carriers of pathogenic/likely pathogenic variants in Lynch syndrome genes to perform prenatal or preimplantation genetic diagnosis [22].
Conclusion
In conclusion, in this study we have identified and characterized as pathogenic aMSH2 exon 15 duplication in a family with Lynch syndrome. CNV analysis in Lynch syndrome patients is of great importance since it can identify pathogenic/likely pathogenic variants with actionable clinical significance. Currently, exonic duplications of MSH2 gene are often classified as VUS because their effect on gene expression is unknown. As in this case, RNA analysis can contribute to the classification of certain exonic duplications in MSH2, as well as in other genes, as pathogenic. As with sequence variants, proper classification of partial MSH2 gene duplications, by performing RNA analysis, is crucial for the management of the patients, since it can lead to targeted therapeutic and risk-reducing interventions, and of their family members, allowing cascade family screening.
Declarations
Ethics approval and consent to participate: The present study was approved by the Ethics Committee of Özel Memorial Antalya Hospital (Antalya, Turkey). All tested individuals provided signed informed consent form before molecular genetic testing and permission for the anonymous use of their data for research purposes and/or scientific publications.
Consent for publication: The proband provided a signed informed consent form prior to molecular genetic testing for the permission of the anonymous use of her data for research purposes and/or scientific publications.
Availability of data and material: All data generated or analyzed during this study are included in this published article. The clinical interpretation of the genomic variant along with the experimental information has been submitted to Clin Var (SUB12060189).
Competing interests: The authors declare that they have no competing interests.
Funding: Not applicable.
Authors’ contributions: DA drafted the manuscript. DA, KA, NT and EP designed the study. DA, KA, GP, and KP carried out the DNA extraction, sequencing and contributed to the analysis and interpretation of the variant data. GNT performed the bio-informatics analysis. MO, OK and MOC provided the materials, demographic data and family history. EP performed the Mismatch Repair (MMR) analysis by immunohistochemistry. GN conceived of the study and participated in its design and coordination.
Acknowledgements: Not applicable.
References
- S. Cox VL, Saeed Bamashmos AA, Foo WC, Gupta S, Yedururi S, Garg N, et al. Lynch Syndrome: Genomics Update and Imaging Review. Radiographics. 2018; 38: 483-499.
- Niessen RC, Hofstra RM, Westers H, Ligtenberg MJ, Kooi K, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009; 48: 737-744.
- Felton KE, Gilchrist DM and Andrew SE. Constitutive deficiency in DNA mismatch repair (Review). Clin Genet. 2007: 71: 483-498.
- Mao R, Krautscheid P, Graham RP, Ganguly A, Shankar S, et al. Genetic testing for inherited colorectal cancer and polyposis, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG).Genet Med. 2021; 23:1807-1817.
- NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment: Colorectal, Version 1.2022. Accessed July4, 2022.http://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf
- Mu W, Li B, Wu S, Chen J, Sain D, et al. Detection of structural variation using target captured next-generation sequencing data for genetic diagnostic testing. Genet Med. 2019; 21: 1603-1610.
- Agiannitopoulos K, Pepe G, Papadopoulou E, Tsaousis GN, Kampouri S, et al. Clinical Utility of Functional RNA Analysis for the Reclassification of Splicing Gene Variants in Hereditary Cancer. Cancer Genomics Proteomics. 2021; 18: 285-294.
- Chen W, Frankel WL: A practical guide to biomarkers for the evaluation of colorectal cancer (Review). Mod Pathol. 2019; 32: 1-15.
- Ye SB, Cheng YK, Zhang L, Zou YF, Chen P: Association of mismatch repair status with survival and response to neoadjuvant chemo(radio)therapy in rectal cancer. NPJ Precis Oncol. 2020; 4: 26.
- Bonadona V, Bonaïti B, Olschwang S, Grandjouan S, Huiart L, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011; 305: 2304-2310.
- Baert-Desurmont S, Buisine MP, Bessenay E, Frerot S, Lovecchio T, et al. Partial duplications of the MSH2 and MLH1 genes in hereditary nonpolyposis colorectal cancer. Eur J Hum Genet. 2007; 15: 383-386.
- National Library of Medicine.Clinvar. Accessed June 15, 2022. https://www.ncbi.nlm.nih.gov/clinvar/
- Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College Of Medical Genetics And Genomics (ACMG) and the Clinical Genome Resource (ClinGen).Genet Med. 2020; 22: 245-247.
- Abou Tayoun AN, Pesaran T, Di Stefano MT, Oza A, Rehm HL. ClinGen Sequence Variant Interpretation Working Group (Clin Gen SVI): Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018; 39: 1517-1524.
- Conner BR, Hernandez F, Souders B, Landrith T, Boland CR, et al. RNA analysis identifies pathogenic duplications in MSH2 in patients with Lynch Syndrome. Gastroenterology. 2019; 156: 1924-1925.
- Roth RM, Haraldsdottir S, Hampel H, Arnold CA, Frankel WL, et al. Discordant Mismatch Repair Protein Immunoreactivity in Lynch Syndrome-Associated Neoplasms: A Recommendation for Screening Synchronous/Metachronous Neoplasms.Am J Clin Pathol. 2016; 146: 50-56.
- Sarode VR, Robinson L. Screening for Lynch Syndrome by Immunohistochemistry of Mismatch Repair Proteins: Significance of Indeterminate Result and Correlation with Mutational Studies.Arch Pathol Lab Med. 2019; 143:1225-1223.
- Yurgelun MB and Hampel H. Recent advances in Lynch Syndrome: Diagnosis, treatment, and cancer prevention. Review Am Soc Clin Oncol Educ Book. 2018; 38:101-109.
- André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004; 350: 2343-2351.
- Tomasello G, Ghidini M, Galassi B, Grossi F, Luciani A, et al. Survival benefit with adjuvant chemotherapy in stage III microsatellite-high/deficient mismatch repair colon cancer: A systematic review and meta-analysis. Sci Rep. 2022; 12: 1055.
- Özdoğan M, Papadopoulou E, Tsoulos N, Tsantikidi A, Mariatou VM, et al. Comprehensive tumor molecular profile analysis in clinical practice.BMC Med Genomics. 2021; 14: 105.
- Daina G, Ramos L, Obradors A, Rius M, Martinez-Pasarell O, et al. First successful double-factor PGD for Lynch syndrome: Monogenic analysis and comprehensive aneuploidy screening. Clin Genet. 2013; 84: 70-73.