
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
A typical bilateral papilledema revealing MOGAD optic
neuritis, with a complete recovery: Case report
Yahya Saoiabi*; Taha Baiz; Hala El Belidi; Salma Hamidi; Nourdine Boutimzine; Lalla Ouafa Cherkaoui
Department of Ophthalmology A, Hospital of Specialties of RABAT, Morocco.
*Corresponding Author : Saoiabi Yahya
Department of Ophthalmology A, Hospital of Specialties of RABAT, Morocco.
Ph: +212 662 88 24 72;
Email: Saoiabi.yahya@gmail.com
Received : Feb 01, 2023
Accepted : Mar 27, 2023
Published : Apr 03, 2023
Archived : www.jcimcr.org
Copyright : © Yahya S (2023).
Abstract
The discovery of papilledema in children constitutes a neuroophthalmological emergency; the purpose of this report is to help differentiate MOGAD from other differential diagnosis of bilateral optic neuritis, especially in childhood.
The 09 years old female came to our emergency department complaining about rapidly progressive visual decline in both eyes, with no other symptom. She has no particular personal or family history. On ophthalmic examination, her BCVA was finger counts OU, with a lazy direct and consensual pupillary reflex in both eyes, and bilateral Frisen grade 2 papilledema on both eyes. Initial blood work was unremarkable, with normal sedimentation rate, C-reactive protein, and negative serologies. MRI with gadolinium contrast revealed an acute posterior bilateral optic neuritis that may be related to an autoimmune or infectious inflammatory origin, and the spinal cord MRI was normal. CSF analysis revealed a normal composition, with a normal LP opening pressure. The diagnosis was made after showing a positive result for the research of anti-MOG antibodies. The main problem is the possibility of recurrence, and that risk is moderate and might be mitigated by prolonged immunosuppression.
The prognosis is generally good, but some complications are not rare (visual, sphincter or cognitive complications) making the knowledge of the symptoms and clinical signs essential for an early diagnosis and optimal management.
Keywords: MOGAD; NMOSD; MS; pediatric papilledema; optic neuritis case report.
Abbreviations: MOGAD: MOG-Igg-Associated Disease; NMOSD: Neuromyelitis Optica Spectrum Disorder; MS: Multiple Sclerosis.
Citation: Saoiabi Y, Baiz T, Belidi HE, Hamidi S, Boutimzine N, et al. A typical bilateral papilledema revealing MOGAD optic neuritis, with a complete recovery: Case report. J Clin Images Med Case Rep. 2023; 4(4): 2354.
Introduction
Papillaryedema is a distension of the intraocular part of the optic nerve, secondary to a blockage of axonal transport at the level of the cribriform plate. This is the case during any attack on the optic nerve, whether the mechanism is ischemic, toxic, inflammatory, compressive, toxic, etc.
The discovery of papilledema in children constitutes a neuroophthalmological emergency, for two reasons: papilledema can lead to optic atrophy; moreover, its cause can jeopardize the vital prognosis of the child.
The purpose of this report is to describe the clinical case of a 9-year-old female with a bilateral papilledema due to anti-MOG NO, it’s ophthalmologic manifestations and treatment.
Case presentation
A 09-year-old female presented to our ophthalmology emergency department with a complaint of 5-day rapidly progressive visual decline in both eyes. She denied pain with ocular movements, headaches, visual field defects, or any other systemic symptoms.
There was no preceding infection or vaccination, no particular ocular or medical history, apart from a notion of exposure to insecticides 1 year ago (mosquito powder used as anti-lice shampoo).
The systemic temperature was 37,2°C. On ophthalmic examination, her Best-Corrected Visual Acuity (BCVA) was finger counts OU.
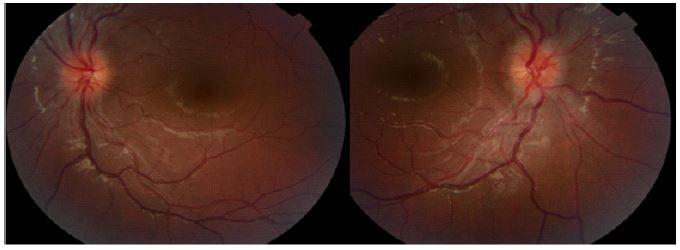
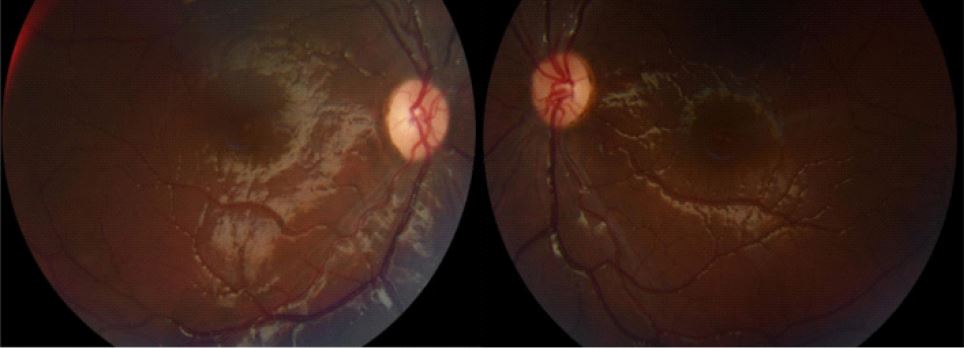
Her direct and consensual pupillary reflex was present but lazy in both eyes, with no relative afferent pupillary defect. All ocular movements were without restrictions and painless. The anterior segment of both eyes was normal on slit lamp examination. Fundoscopy revealed bilateral Frisen grade 2 papilledema on both eyes, hypereamia papilla with blurred contours, dilated veins, and filling of the excavation.
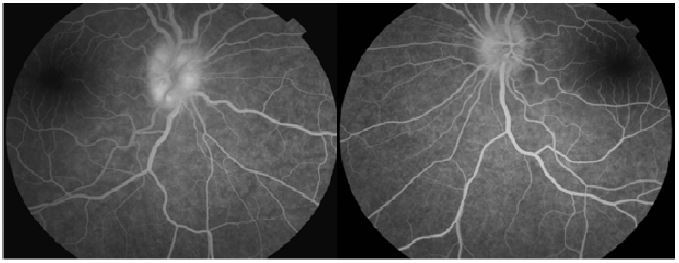
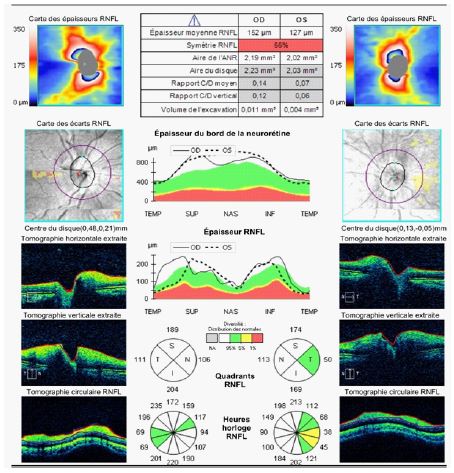
There was no hemorrhages or exudates, no macular edema, no vasculitis, no hyalitis. OCT confirmed the fundoscopy findings. Humphrey visual field wasn’t reliable and showed lot of false negatives and positives.
Initial blood work was unremarkable, with normal values of white blood cell count, thrombocytes, sedimentation rate, C-reactive protein, and negative serologies (Syphilis, toxoplasmosis, tuberculosis, CMV, HSV, HIV, EBV, HVB and HVC).
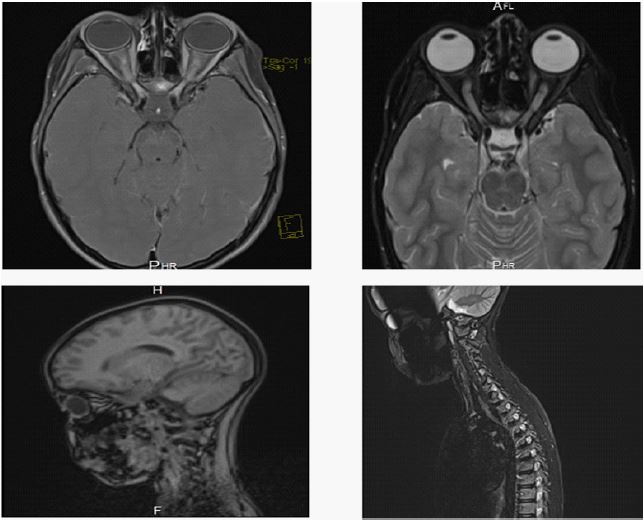
Magnetic Resonance Imaging (MRI) with gadolinium contrast was then performed (without evidence of mass lesion, hydrocephalus, or meningeal enhancement) it revealed an acute posterior bilateral optic neuritis that may be related to an autoimmune or infectious inflammatory origin, and the spinal cord MRI was normal.
Cerebrospinal Fluid (CSF) analysis revealed a normal composition, with a normal LP opening pressure.
Intravenous methylprednisolone pulse therapy (10 mg/kg/day for 05 consecutive days; IVMP) relayed by oral steroids markedly decreased symptoms, with improvement of visual acuity (4/10 OU on the 8th day of treatment, 10/10 OU 1 month later).
An internationally strandardized Cell-Based Assay (CBA) for Anti-bodies anti MOG and AQ4 showed a positive anti MOG, and a negative anti AQ4.
The diagnosis of anti MOG demyelinating inflammatory optic neuritis without transverse myelitis was then been made.
Discussion
A demyelinating neurologic disease in the CNS is called Neuromyelitis Optica Spectrum Disorder (NMOSD). NMOSD is characterized by the presence of particular antibodies such as Anti-Aquaporin-4 Immunoglobulin G (AQP4-IgG) and Anti-Myelin Oligodendrocyte Glycoprotein Immunoglobulin G (MOG-IgG), in contrast to Multiple Sclerosis (MS), which typically lacks disease-specific antibodies.
Optic Neuritis (ON) and acute myelitis are the most common recurrent neurologic episodes seen in individuals with MOG-IgG-associated disease and those with AQP4-IgG-positive NMOSD. Although, these 2 diseases present similar clinical manifestations in the acute attack, but the sequelae are generally worse in AQP4-igG-positive NMOSD [1,2], arguing that these 2 diseases should be viewed as separate disease entities with various relapse prevention strategies [3].
That’s why recently; we start to consider the MOG-IgG-associated disease as a separate entity, called MOGAD (MOG Associated Disease).
Compared to MS, NMOSD attacks are typically more severe and recover less quickly.
According to research, compared to 4% of MS ON patients after a 15-year follow-up, 60% of NMOSD patients had unilateral or bilateral blindness with median disease duration of 7.7 years [4].
Optical Coherence Tomography (OCT) studies have revealed that the retinal nerve fiber layer thinning is greater in NMOSD patients with ON compared with MS patients [5].
Inflammatory lesions affecting the central gray matter of the spinal cord can appear on MRI in people with NMOSD, extending over three or more adjacent vertebral segments [6].
The timing of the MRI determines how long the lesion will last because a signal anomaly may disappear or shorten over time.
In opposition to MS, recovery from attacks is typically partial, and patients gradually experience attack-related dysfunction. Spotty lesions that have cavitation and central necrosis are possible. More than half of all spinal cord injuries take up more than half of the cross section of the spinal cord [7].
Short lesions should not rule out the diagnosis of NMO even though long lesions are more common. According to a recent study, 14% of patients have small lesions visible on MRI [8].
The principal distinction between MOG-antibody and AQP4-antibody diseases, is that MOG-antibody disease is considered milder, and fortuitously less relapsing [3,9].
Anti-MOG antibodies are directed towards oligodendrocytes and cause acute demyelinating lesions with good recovery potential. Conversely, anti-AQP4 antibodies are directed towards astrocytes, the destruction that ends up in lesions with a poorer prognosis.
MOGAD can present at any age, most typically with atypical optic neuritis, with a slight female predominance and no ethnic bias.
Optic Neuritis (ON) is the most common initial presentation of MOGAD in adolescence and adulthood, and a frequent presentation in pediatric patients [10,11].
Up to 80% of patients have bilateral optic nerve involvement, which is quite uncommon in MS, and visual loss is frequently severe at the time of onset [12]. Even though the acute period of visual loss is severe, recovery is typically good, especially in youngsters [12].
Optic disc edema is uncommon in MS or NMOSD, however it can occur in up to 86% of MOGAD-ON patients [13-15].
MOGAD frequently manifests as Acute Disseminated Encephalomyelitis (ADEM) or a condition similar to ADEM in young children. (ADEM with optic neuritis, multiphasic disseminated encephalomyelitis) [11,16], large, poorly defined bilateral lesions usually involving cortical and deep gray matter areas are typical findings on brain MRIs [17].
Particularly indicative of MOGAD are episodes of recurrent ADEM or ADEM coupled with recurrent optic neuritis. Importantly, even when MRI results are not consistent with ADEM in children with clinical presentation of encephalitis, MOGAD diagnosis is still possible [18].
Over 90% of MOGAD cases progress in a relapsing manner [19], with the risk of recurrence influenced by the duration of immunosuppression initiated after the onset attack.
Relapses of transverse myelitis or optic neuritis typically start out slowly over a few days before improving over the course of several weeks or months [20].
In contrast to MS, when disability typically occurs as part of the illness’s progressive phase, disability is typically attack-related [21,22].
The prognosis is typically favorable; however, patients can be left with significant sphincter and erectile dysfunction, cognitive impairment and poor visual acuity. The majority of this disability originates from the onset attack.
The first-line therapy is based on intravenous steroids with or without oral relay, and the prescription of intravenous immunoglobulin and or plasmapheresis in the event of poor recovery of visual acuity.
The “main treatment” is based on immunosuppressants, and for the most severe cases the administration of rituximab [23].
Autoantigen-based treatments provide the promise of combining a high efficacy with a good tolerability because they should only interfere with pathogenic immune responses and leaving the rest of the immune system unaffected [24].
Conclusion
Optic neuritis is a pathology frequently encountered in ophthalmology; a good knowledge of the symptoms and clinical signs is essential for an early diagnosis and optimal management.
MOG-antibody disease is a newly identified central nervous system inflammatory disease, which is confused with multiple sclerosis but distinct from it, and is responsible of attacks involving the optic nerve, spinal cord, brainstem and the brain.
The problem is the possibility of recurrence, and that risk is moderate and might be mitigated by prolonged immunosuppression.
The prognosis is generally good, except for a certain number of patients, who may end up with visual, sphincter or cognitive complications.
Declarations
Competing interests: Absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors’ contributions: All authors contributed to manuscript revision, read and approved the submitted version.
References
- Akaishi T, Nakashima I, Takeshita T, et al. Different etiologies and prognoses of optic neuritis in demyelinating diseases. J Neuroimmunol. 2016; 299: 152-157.
- Liu H, Zhou H, Wang J, et al. The prevalence and prognostic value of myelin oligodendrocyte glycoprotein antibody in adult optic neuritis. J Neurol Sci. 2019; 396: 225-231.
- Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: A comparative study. JAMA Neurol. 2014; 71: 276-283.
- Visual function 15 years after optic neuritis: A final follow-up report from the Optic Neuritis Treatment Trial. Ophthalmology. 2008; 115: 1079–1082e5.
- Green AJ, Cree BA. Distinctive retinal nerve fibre layer and vascular changes in neuromyelitis optica following optic neuritis. J Neurol Neurosurg Psychiatry. 2009; 80: 1002–1005.
- Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG, et al. The spectrum of neuromyelitis optica. Lancet Neurol. 2007; 6: 805–815.
- Nakamura M, Miyazawa I, Fujihara K, Nakashima I, Misu T, Watanabe S, et al. Preferential spinal central gray matter involvement in neuromyelitis optica. An MRI study. J Neurol. 2008; 255: 163–170.
- Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, et al. Short Myelitis Lesions in Aquaporin-4-IgG-Positive Neuromyelitis Optica Spectrum Disorders. JAMA Neurol. 2014.
- Doulglas Kazutoshi Sato et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014; 82: 474481.
- Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al.. Clinical presentation and prognosis in MOG-antibody disease: A UK study. Brain. 2017; 140: 3128–3138.
- Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. 2017 ; 89: 900–908.
- Chen Q, Zhao G, Huang Y, Li Z, Sun X, Lu P, et al. Clinical characteristics of pediatric optic neuritis with myelin oligodendrocyte glycoprotein seropositive: A cohort study. Pediatric Neurol. 2018; 83: 42–49.
- Chen JJ, Flanagan EP, Jitprapaikulsan J, López-Chiriboga AS, Fryer JP, Leavitt JA, et al. Myelin oligodendrocyte glycoprotein antibody–positive optic neuritis: Clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018; 195: 8–15.
- Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. MultSclero J. 2016; 22: 470–482.
- Zhao Y, Tan S, Chan TC, Xu Q, Zhao J, Teng D, et al. Clinical features of demyelinating optic neuritis with seropositive myelin oligodendrocyte glycoprotein antibody in Chinese patients. British J Ophthalmol. 2018; 102: 1372–1377.
- Pröbstel AK, Dornmair K, Bittner R, Sperl P, Jenne D, Magalhaes S, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology. 2011; 77: 580–588.
- Wegener-Panzer A, Cleaveland R, Wendel EM, Baumann M, Bertolini A, Häusler M, et al. Clinical and imaging features of children with autoimmune encephalitis and MOG antibodies. Neurol Neuroimmunol Neuroinflamm. 2020; 7: e731.
- Armangue T, Olivé Cirera G, Martínez Hernandez E, Sepulveda M, Ruiz Garcia R, Muñoz Batista M, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: A multicentre observational study. Lancet Neurol. 2020; 19: 234–246.
- Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG, et al. The spectrum of neuromyelitis optica. Lancet Neurol. 2007; 6: 805–881.
- Mealy MA, Wingerchuk DM, Greenberg BM, Levy M, et al. Epidemiology of neuromyelitis optica in the United States: A multicenter analysis. Arch Neurol. 2012; 69: 1176–1180.
- Jiao Y, Fryer JP, Lennon VA, Jenkins SM, Quek AM, Smith CY, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013; 81: 1197–1204.
- Collongues N, Cabre P, Marignier R, Zephir H, Papeix C, Audoin B, et al. A benign form of neuromyelitis optica: Does it exist? Arch Neurol. 2011; 68: 918–924.
- Kinzel S, Weber MS. The Role of Peripheral CNS-Directed Antibodies in Promoting Inflammatory CNS Demyelination. Brain Sci. 2017; 7: 7. http://dx.doi.org/10.3390/brainsci7070070
- Derfuss T, Meinl E. Identifying autoantigens in demyelinating diseases: Valuable clues to diagnosis and treatment? Curr Opin Neurol. 2012; 25: 231-238.