
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
A rare presentation in Von Hippel–Lindau
syndrome: Teratoma of testis
Zhongjian Li; Liankai Zhu; Shaokang Du; Zhibo Wang; Jintao Chen; Huajun Zhang; Xuwei Zhao; Hongyao Liu*; Xiaochun Liu*
Department of Urology, Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Taiyuan, 030032, China.
*Corresponding Author : Liu &
Xiaochun Liu
Department of Urology, Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Taiyuan, 030032, China.
Email: zhanghuajun0705@163.com;
zhaoxuwei@163.com
Received : Feb 07, 2023
Accepted : Apr 04, 2023
Published : Apr 11, 2023
Archived : www.jcimcr.org
Copyright : © Liu H & Liu X (2023).
Abstract
Von Hippel-Lindau Syndrome (VHL) is a rare genetic disease. The clinical and imaging data of one patient with VHL syndrome were retrospectively analyzed, and the clinical features and treatment of the disease were analyzed in combination with literature. The patient had multiple cysts of the pancreas, multiple cysts of both kidneys, left kidney tumor, right testicular tumor, and bilateral epididymal cystic lesions, and underwent surgery for right orchiectomy, and was discharged after surgery. The possibility of VHL syndrome should be considered for tumors involving the central nervous system, pancreas, kidneys and other systems, and when the lesion involves the scrotum, not only the papillary cystadenoma of the epididymis, surgical resection should be the preferred treatment when malignancy is suspected.
Keywords: VHL syndrome; Testicular tumor; Diagnosis; Treatment.
Citation: Li Z, Zhu L, Du S, Liu H, Liu X, et al. A rare presentation in Von Hippel–Lindau syndrome: Teratoma of testis. J Clin Images Med Case Rep. 2023; 4(4): 2365.
Background
Von Hippel-Lindau (VHL) syndrome is a rare clinical familial autosomal dominant tumor syndrome involving multiple body systems. Patients with VHL may develop multiple benign and malignant tumors involving various organ systems, including retinal Hemangioblastomas (HBs), Central Nervous System (CNS) HBs, endolymphatic sac tumors, pancreatic neuroendocrine tumors, pancreatic cystadenomas, pancreatic cysts, clear cell renal cell carcinomas, renal cysts, pheochromocytomas, paragangliomas, and epididymal and broad ligament cystadenomas. Diagnosis of VHL is based on positive family history of VHL disease with one associated tumor. The present report describes a patient with VHL syndrome who was treated with testectomy.
Clinical data
A 33 years old man, a known case of VHL syndrome, has a palpable mass in the right testis. In the past 10 years, the patient “cerebellar angioreticuloma” has recurred many times, all undergone surgical treatment. He was diagnosed with VHL disease in 9 years ago. One month ago, he was hospitalized for radiotherapy in our hospital because of recurrent cerebellar hemangioblastoma. The computed tomography shows that he has multiple cysts of the pancreas, multiple cysts of both kidneys, left kidney tumors, right testicular tumors and bilateral epididymal cystic lesions. Physical examination shows the right epididymis is enlarged, about 3*3 cm, the scrotal skin is normal, the right testicle is larger than the left, the hard mass of about 4*4 cm can be palpated, and no tenderness. His father died of cerebellar angioblastoma and his Aunt’s little son died of cerebellar angioblastoma. Laboratoryalpha-fetoprotein, human chorionic gonadotropin, luteinizing hormone, follicle-stimulating hormone, serum testosterone were normal. Abdominal ultrasound indicate that diffuse increase in the volume of the pancreas with multiple cystic lesions. The middle and upper cystic lesions of the left kidney are not excluded, and multiple cysts in the right kidney are not excluded. Abdominal and pelvic CT display diffuse multiple cysts of the pancreas with calcification, multiple cysts of both kidneys, malignancy of the left kidney, multilocular cystic lesions of the right testis with abnormal strengthening, and malignant mass of the testicles. Right-sided orchiectomy was performed after completing relevant examinations, and postoperative pathological diagnosis: right testicles: cystic lesions, and monodermal teratoma (goiter) with papillary thyroid carcinoma was considered. The patient treated kidney cancer with oral sunitinib malate after surgery, but after 4 weeks of oral administration, the patient developed side effects of limb weakness and loss of appetite, and stopped the drug on his own. Half a year after discharge, the urinary ultrasound was re-examined, and there was no significant progress in both kidney lesions, and no recurrence of testicular tumors. At present, the patient is bedridden, with weakness in limbs, numbness in the left hand, poor hearing and vision. Occasionally nausea, vomiting, drinking water and coughing.
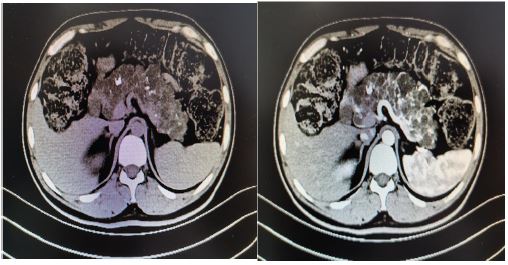
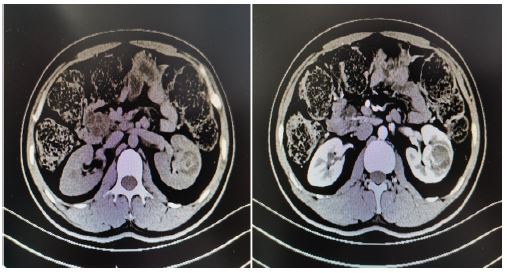
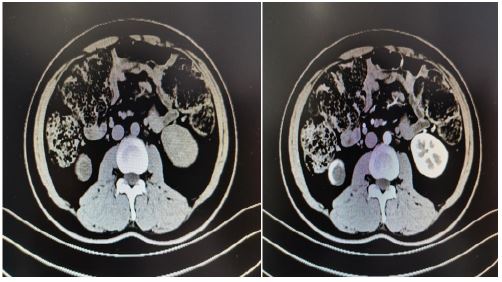
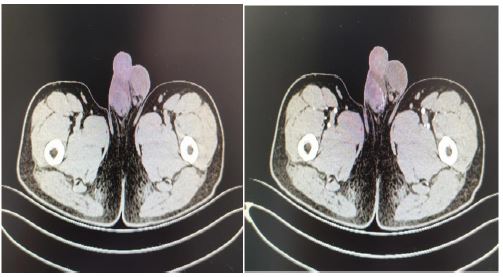
Discussion
Von Hippel-Lindau Disease (VHL) is a rare autosomal dominant disorder caused by a mutation in the tumor suppressor gene at 3p25-26, affecting 1 in 35 000-40 000 of the population [1]. In 1964, Melmon and Rosen first officially named central nervous system hemangiocytoma with kidney or pancreatic cysts, pheochromocytomas, kidney cancer, and epithelial cystadenomas “Von Hippel-Lindau syndrome”, or VHL syndrome for short. Systemic or germ line mutations in the VHL gene cause multiple organs to develop benign, malignancies, and cysts, including benign and malignant tumors in several systems of the Central Nervous System (CNS) and internal organs such as the kidneys, pancreas, adrenal glands, and reproductive organs. The most common VHL-associated tumors are angioblastomas involving the cerebellum, spinal cord, and retina, clear cell carcinomas of the kidney, pheochromocytomas and paragangliomas, and pancreatic neuroendocrine tumors.
The molecular basis of VHL syndrome is that mutations in the VHL gene located on the short arm of chromosome 3 (3p25-26) cause impaired VHL protein synthesis [2]. Most cases of VHL syndrome are familial, but sporadic in 20 percent of cases are caused by new mutations [3]. Missense mutations are detected in 30% to 60% of VHLS cases; Large fragment deletions from 0.5 kb to 250 kb account for 20% to 40% of genetic changes, about 12% to 20% are microdeletions or insertions, and nonsense mutations account for 7% to 11% of cases. Mutations can be located throughout the coding region of the gene [2,3]. Analysis of VHL gene mutations showed correlation with its genotype/phenotype, and mutations leading to early termination of proteins (meaninglessness, insertions, and deletions) were most common in VHL cases without pheochromocytoma. Based on the type of gene mutation, VHL disease can be divided into 2 types, type 1 VHL mutations are mainly a large number of deletions or truncation, resulting in low or no activity of the encoded protein, it is related to retinal and central angioblastoma and renal clear cell carcinoma, and the probability of pheochromocytoma is low. Type 1A, with renal cell carcinoma. Type 1B, without renal cell carcinoma. Missense mutations are most common in type 2 VHL, and clinical concomitant pheochromocytoma is common. Type 2A pheochromocytoma, retinal and central nervous system angioblastoma, no renal cell carcinoma; Type 2B pheochromocytoma, retinal and central nervous system angioblastoma, pancreatic cyst and neuroendocrine tumor, renal cell carcinoma; Type 2C only pheochromocytoma. In addition, patients with VHL may have polycythemia, but patients with VHL-related tumors may not have congenital polycythemia.
VHL syndrome has a penetrance rate of more than 90 percent in people 65 years of age [5], early manifestations occur around the age of 20, and life expectancy in VHL patients previously ranged from 40 to 52 years, averaging 59.4 years in men and 48.4 years in women [6]. The most common causes of death are angioblastoma and metastatic kidney cancer of the central nervous system and their complications. This study reported in 1 patient with VHL syndrome with cerebellar angioblastoma-containing cerebellar angioblastoma, left kidney tumor, double kidney cyst, and right testicular tumor. Diagnosis and classification of VHL syndrome: Maher et al. [2] proposed imaging diagnostic criteria for VHL syndrome: (1) more than two central nervous system hemangioblastoma or one central nervous system angioblastoma plus retinal angioblastoma; (2) An angioblastoma of the central nervous system plus abdominal organ lesions (mainly refers to one side of multicentric or bilateral kidney cancer, pheochromocytoma, visceral cysts such as kidney, adrenal gland, pancreas, liver, mesentery, omentum cyst, liver, epididymal adenoma, etc.); [3] Have a positive family history plus an angioblastoma or any abdominal organ lesion.
Renal manifestations of VHL include benign renal cysts and clear cell carcinoma. Clear cell renal carcinoma manifestations appear in 70% of VHL patients up to 60 years of age [4]. Renal cysts are usually asymptomatic, and surgical intervention is not recommended for renal cysts without RCC, and surgery is the only treatment for cysts with pain or compression. Kidney cancer may affect up to 30% of VHL patients, who develop renal cell carcinoma and renal cysts at the age of 30-40 years, which are rarely the first symptoms of VHL. Up to 70 percent of patients with VHL develop renal cell carcinoma before the age of 60 years, and renal cell carcinoma is the leading cause of death in these patients. VHL nephropathy ranges from simple cystic to complete solid. Clinical manifestations include a renal mass with low back pain or hematuria. Simple renal cysts are generally asymptomatic, while complex cysts become substantial renal masses. Renal function is preserved despite multiple cysts. RCC is usually detected during routine screening and does not require intervention for tumors smaller than 3 centimeters in diameter. For tumors that reach the 3 cm threshold, partial nephrectomy is the treatment of choice to reduce the risk of metastasis while preserving renal function. Nephron-sparing surgery has a 10-year cancer survival rate of up to 81 percent with renal function preserved for lesions larger than 3 centimeters, and percutaneous and laparoscopic radiofrequency ablation has been shown to be effective for smaller tumors (< 3 centimeters) with low complication rates [7]. However, radiofrequency ablation requires frequent monitoring and intervention. In this case, the left kidney mass was not treated with surgery, and sunitinib malate was given oral 4/2 regimen, but after 4 weeks of oral administration, the patient developed limb weakness and side effects of loss of appetite, and stopped the drug on his own. Six months after discharge, the urinary ultrasound was re-examined, and there was no significant progress in both kidney lesions.
Men with VHL can develop epididymal cystadenoma. They can occur in 25-60% of male patients with VHL, unilaterally or bilaterally. They are benign in nature and do not require surgery [8]. These tumors usually appear in the teenage years. They are usually asymptomatic and are discovered incidentally. In 17 percent of patients, lesions cannot be palpated due to their small size. Ultrasonography is the preferred method of localization of these lesions. The morphological characteristics of this tumor are similar to those of other VHL tumors. Diagnosis of epididymal cystadenoma is confirmed by palpation and ultrasound. Due to their small size, these lesions are difficult to palpate; Therefore, ultrasonography is the preferred test method. Combined with physical examination and auxiliary examination, the patient considered the right testicular mass and underwent right orchiectomy, postoperative pathology considered the cystic lesion of the right epididymis, the right testicle contained red stained gum samples, some areas of the epithelium were papillary hyperplasia, cytoplasm was transparent, the nucleus was atypic, and the monodermal teratoma (goiter) with papillary thyroid carcinoma was considered.
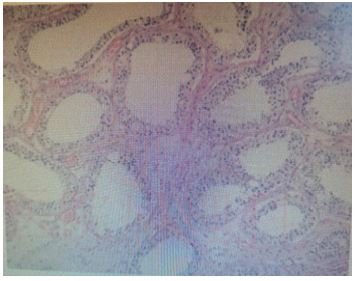
Testicular tumors are rare, occurring in young and middle-aged men, accounting for 1 percent of all male tumors. More than 95% of testicular cancers come from germ cells. Germ cell testicular cancers are divided into seminomas (classical, anaphylactic, and spermatocytomic) and nonseminomas (embryonal carcinoma, teratoma, teratoma, choriocarcinoma, and yolk sac tumors). Nongerm cell tumors include sex cord-gonadal stromal tumors (derived from mesenchymal cells or supporting cells) and other tumors, and testicular tumors are mostly germ cell tumors. In general, testicular secondary tumors are uncommon, and the scrotal environment is not suitable for metastasis due to scrotal temperatures lower than elsewhere in the body and the presence of a blood-testicular barrier formed by supporting cells to protect sperm [9].
Testicular teratomas typically present as a single, painless mass in the scrotum on the affected side, occasionally accompanied by dull pain in the scrotum or discomfort in the lower abdomen. Both physical examination and imaging are similar to testicular tumors. Serum tumor markers play an important role in diagnosis, staging, and prognosis. AFP, HCG, and lactate dehydrogenase are predominantly included and lack sensitivity and specificity, and these assays are normal. Ultrasonography is the preferred examination method for testicular tumors, and is mostly used for testicular tumor screening and early diagnosis. It is often manifested as uneven echo of the mass, containing liquid dark areas of varying sizes or strong echoic foci. The mass is attached to hair, teeth or bones. Some teratomas are solid lobulations or predominantly solid masses containing many small cystic cavities, and immature bone or cartilage tissue is visible in the solid area. CT of testicular teratoma is characterized by soft tissue density mass and fat density shadow with calcification points in the testicular parenchymal, and if the teratoma mass is blurred or absent from the surrounding fat space, and peripheral infiltrates are found, a high degree of vigilance for testicular teratoma malignancy is found. This patient mainly presents with a single painless hard mass in the right scrotum, and for patients with VHL syndrome, it is common to have central nervous system or retinal tumors, pancreatic or renal cysts and tumors, epididymal cyst adenomas, and most testicular tumors are kidney cancer metastasizing to the testicles. The testicular pathological manifestations of this patient were considered to be the special lesions of VHL syndrome in the testicles, which were not reported in the literature. No testicular tumor recurrence was seen at follow-up for six months after surgery.
Conclusion
Clinicians need to raise awareness of VHL syndrome when multiple tumors such as central nervous system or retinal tumors, pancreas, or kidneys are found to be present in patients with central nervous system or retinal tumors, pancreas, or kidneys, and benign or malignant lesions containing the testicles should be considered when lesions occur in the scrotal area of patients with VHL, in addition to the common papillary cystadenoma of the epididymis. More evaluation is needed for early detection and treatment of possible metastases. If metastasis and malignancy cannot be definitively excluded, radical orchiectomy and histopathological examination are recommended.
References
- Fatih Turgut N, Crunkhorn R, Iqbal J, Dasgupta S. Vestibular Function in Children With Von Hippel-Lindau Disease. J Int Adv Otol. 2021; 17: 361-367.
- Maher E, Neumann H, Richard S. Von Hippel-Lin-dau disease: A clinical and scientific review. European Journal of Human Genetics. 2011; 19 : 617–623.
- Varshney N, Kebede A, Owusu-Dapaah H, Lather J, Kau-shik M, Bhullar J. A review of Von Hippel-Lindau syndrome. Journal of Kidney Cancer and VHL. 2017; 4: 20–29.
- Varshney N, Kebede AA, Owusu-Dapaah H, Lather J, Kaushik M, Bhullar JS, et al. A review of V on Hippel-Lindau syndrome. J Kidney Cancer VHL. 2017; 4: 20-29.
- KARIMI S, ARABI A, SHAHRAKI T, et al. Von Hippel-Lindau disease and the eye [J]. J Ophthalmic Vis Res. 2020; 15: 78.
- Wilding A, Ingham SL, Lalloo F, Clancy T, Huson SM, Moran A,et al. Life expectancy in hereditary cancer predisposing diseases: An observational study. J Med Genet. 2012; 49: 264–269.
- Young EE, Castle SM, Gorbatiy V, Leveillee RJ. Comparison of safety, renal function outcomes and efficacy of laparoscopic and percutaneous radio frequency ablation of renal masses. J Urol. 2012; 187: 1177–1182.
- Chittiboina P, Lonser RR. Von Hippel–Lindau disease. Handb Clin Neurol. 2015; 132: 139–156.
- Nouralizadeh A, Afyouni A, Shakiba B, Radhi FK. Simultaneous bilateral laparoscopic adrenalectomy for adrenal metastases of renal cell carcinoma: A case report. J Endourol Case Rep. 2017; 3: 142–145.