
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Impact of genomic characterization in patients
with spinal muscular atrophy no 5q
Moreno-Giraldo Lina Johanna1; Ponce-Ramírez María Alejandra2*
11MD, Pediatrician. MSc. and PhD. in Medical Genetics, Postgraduate in clinical bioinformatics, attached to Universidad del Valle, Universidad Santiago de Cali, Universidad Libre.
22MD, attached to Universidad Santiago de Cali Seccional Palmira, Colombia.
*Corresponding Author : Ponce-Ramírez MA
Universidad Santiago de Cali Seccional Palmira, Colombia.
Email: Alejandraponce15@outlook.es
Received : Feb 06, 2023
Accepted : Apr 07, 2023
Published : Apr 14, 2023
Archived : www.jcimcr.org
Copyright : © Alejandra PRM (2023).
Abstract
Introduction: Spinal Muscular Atrophy (SMA), is defined as a set of hereditary neurodegenerative disorders causing a high phenotypic and genotypic variability that generate an impact on quality of life, psychosocial, emotional and functional development, considered in Colombia orphan disease in relation to its low prevalence, chronicity and high complexity.
Objective: To describe, characterize and correlate phenotypically and genotypically a patient with clinical suspicion of neurodegenerative disease.
Clinical case: A 32-year-old female patient with clinical picture consisting of equinismus, varus, hindfoot supination, right forefoot adduction and limitation in wrist extension with subsequent weakness and muscle atrophy predominantly in the lower limbs, generalized areflexia and positive Gowers sign, with suspicion of progressive degenerative neuromuscular disease, endocrine, neuromuscular, cardiovascular studies, sural nerve biopsy and genetic study were requested.
Results: Biopsy of sural nerve with loss of axons with little demyelination, hypertrophy of Schwann cells, and genomic study clinical exome sequencing trio performed using Illumina technology with identification of variants with pathogenic clinical significance in the NOD2 gene with heterozygous zygosity and homozygous DYNC2H1, finally gene interaction network is performed using Gene Mania program determining gene associations.
Conclusion: The diagnosis of SMA represents a challenge due to its wide phenotypic-genotypic variability, although most patients are due to variants in the SMN1 gene there are other non-5q genes associated with this pathology, a specific diagnosis impacts on treatment, prognosis and morbimortality attributed, establishing heritability risk and genetic counseling for the sake of preventive, predictive, personalized and participatory medicine.
Keywords: Spinal Muscular Atrophy; Orphan disease; Neuromuscular; Genetic characterization; Correlation; Preventive medicine.
Citation: Moreno-Giraldo LJ, Ponce-Ramírez MA. Impact of genomic characterization in patients with spinal muscular atrophy no 5q. J Clin Images Med Case Rep. 2023; 4(4): 2372.
Introduction
Spinal Muscular Atrophy (SMA) is defined as a group of hereditary neurodegenerative disorders, capable of causing an alteration in the cells of the anterior horn in the spinal cord and the motor nuclei in the lower part of the brainstem, causing progressive symmetrical muscle weakness and atrophy whose severity depends on the age of onset, in addition to chronically debilitating long-term complications that threaten the lives of patients [1-4].
Resolution 023 of 2023 “Whereby the list of orphan - rare diseases in Colombia is updated” includes SMA as an orphan disease, due to its low prevalence, chronicity and high complexity; it is classified into SMA associated with a genetic variant in the long arm of chromosome 5 (SMA 5q) and non-5q SMA [2].
SMA 5q represents 80-90% of hereditary motor neuron disorders, with an incidence of 1 in 6000-10000 live births, secondary to deletion of the motor neuron Survival Gene (SMN1) located on chromosome 5q11.2-q13. 3, telomeric, considered as the determinant of the disease since its absence or genetic variants constitute the genetic confirmation of the clinical suspicion and the motor neuron Survival Protein 2 (SMN2) gene, centromere, homologous to SMN1, considered a phenotypic modifier [5,6].
In Colombia, according to the National Institute of Health, in an epidemiological period from 2016 to 2020, 2,159 cases of diseases of the nervous system were reported, among which 3 cases of other unspecified SMA were identified, representing 0.04% of the 8,555 cases of orphan-rare diseases. 555 cases of orphan-rare diseases [7], during 2021 the behavior of nervous system diseases reported by the Public Health Surveillance System (SIVIGILA) was 1,187 cases of the 4,949 orphan diseases. 949 orphan diseases, among which they do not describe which diseases correspond to each case [8], in relation to the epidemiological bulletin of 2022, 1,368 cases of nervous system diseases were reported to SIVIGILA with the identification of 9 cases of other unspecified SMA representing 0.14% of the 6,657 cases of orphan diseases [30].
The number of causative genes associated with non-5q SMA has expanded rapidly due to the advent of next-generation sequencing technologies, they are usually classified genetically according to autosomal dominant, autosomal recessive or X-linked inheritance pattern and distribution of proximal, distal or bulbar muscle weakness [9-11] (See Table 1 and 2).
Pathophysiological mechanisms secondary to motor neuron damage include abnormalities in DNA deoxyribonucleic acid) repair (UBA1), altered RNA (ribonucleic acid) processing and degradation (EXOSC3, EXOSC8), vitamin uptake (SLC52A3 and SLC52A2), cell cycle regulation (VRK1, TFG), lipid metabolism (ASAH1), nuclear transport (GLE1), regulation of autophagy control, cytoskeleton dynamics and RNA metabolism (LAS1L, IGHMBP2, AR, SETX), molecular transport by cation channeling (TRPV4), nuclear transport (LMNA), nuclear transcription (AR), axonal transport (BICD2 and DYNC1H1), immune and inflammatory regulation (TFG), ganglioside accumulation in the lysosome (HEXB), structural and functional abnormalities of mitochondria (CHCHD10) [9,15].
The clinical spectrum of SMA is broad and heterogeneous, the clinical presentation of non-5q SMA depends on each genetic variant and the areas involved, the main clinical manifestation of lower motor neuron involvement is muscle weakness (proximal or distal, symmetrical or asymmetrical, in upper or lower limb and with or without involvement of the respiratory musculature), if there is any involvement in bulbar motor neurons, swallowing and voice disorders will be present and finally hyporeflexia or areflexia, The clinical presentation of some types of non-5q SMA can be similar to 5q SMA in which there is evidence of fetal hypomotility, muscle weakness, hypotonia, areflexia, facial diplegia, arthrogryposis or respiratory insufficiency, however, the complementary study, treatment and genetic counseling are clearly different [2,4,14,16].
SMA is considered a medical challenge, the diagnostic suspicion begins when confronted with a patient with clinical manifestations consisting of muscle weakness or hypotonia not attributed to secondary causes; clinical signs suggesting lower motor neuron involvement (muscle atrophy, fasciculations, hypotonia, clubfoot, deforming contractures, winged scapula, hypoarreflexia or areflexia), the age of onset of clinical manifestations considering that there are pictures of rapid or slow progression, the clinical heterogeneity according to the early (congenital, early infancy, early childhood) or late (young adult, older adult) onset of symptoms, the search for involvement of other systems or organs, and the knowledge of the different entities described in Table 1 and 2, which will allow making the right decisions in order to have a specific diagnosis [2,3].
Confirmation of the diagnosis is made by molecular genetic testing with analysis of specific genetic variants that detect homozygous deletions of exons 7 of SMN1, the most common genetic variant in SMA, however, due to the wide genetic variability of non-5q SMA, obtaining the presence of the SMN1 genetic variant with non-pathogenic clinical significance does not rule out the presentation of SMA; Electromyography and muscle biopsy were previously a standard part of the diagnostic evaluation of SMA, however, molecular genetic testing is now widely available [2,3].
Currently there is no established treatment for each of the non-5q SMAs, according to Clinical Trials 215 SMA-related studies have been documented from 2000 to the present year, including two studies associated with non-5q SMA, a Phase I/IIa intrathecal gene delivery clinical trial for IGHMBP2 gene-related disease with an estimated study completion date of November 2028; trial to evaluate the long-term safety and efficacy of leuprorelin acetate injection kit 11.25 mg in patients with spinal and bulbar muscular atrophy in the routine clinical setting with an estimated study completion date of August 31, 2025; meanwhile in the literature Lee J, Termglinchan V, Diecke S et al. [20] describe a study in which a Lamin A/C (LMNA)-dilated cardiomyopathy model was performed in vitro using patient-specific induced pluripotent Pluripotent Stem Cell-Derived Cardiomyocytes (iPSC- CM) in which pharmacological and molecular inhibition of the Platelet-Derived Growth Factor (PDGF) signaling pathway was shown to ameliorate the arrhythmia phenotypes of mutant iPSC-CM in vitro suggesting PDGF receptor beta as a potential therapeutic target, However, adequate dosing or alternatives to these inhibitors have not been established by clinical trials; Therefore, the therapeutic management of patients with non-5q SMA is limited to orthopedic, nutritional, ventilatory and rehabilitative support therapies according to the clinical presentation of the patient [1,17-20].
Table 1: Genotypic and phenotypic characteristics associated with the presentation of early-onset non-5q SMA.
| Gene | OMIM | Name | Location chromosomal | Function | Clinic and onset of symptoms | Heritage | Condition | |
|---|---|---|---|---|---|---|---|---|
| EARLY ONSET | ||||||||
| SMAX2 | UBA1 | 314370 | Ubiquitin-like modifier that activates enzyme 1. | Xp11.3 | Catalyze first step in ubiquitin conjugation | Antenatal. Hypotonia, areflexia, thoracic deformities, facial dysmorphic features, joint contractures, bone fractures, genital anomalies. | X-linked recessive | Lethal infantile spinal muscular atrophy with arthrogryposis, congenital fractures |
| BVVLS1 | SLC52A3 | 613350 | Solute transporter family 52 member 3 | 20p1.3 | Encoding riboflavin transporter protein | Early childhood-adulthood. Weakness of arms, hands and face; ataxia; dysphagia; lingual atrophy, pontobulbar palsy, sensorineural deafness. | Autosomal recessive | Brown Vialetto-Van Laere syndrome 1 |
| BVVLS2 | SCL52A2 | 613350 | Solute transporter family 52 member 2 | 8q24.3 | Encoding riboflavin transporter protein | Early childhood-adulthood. Weakness of arms, hands and face; ataxia; dysphagia; lingual atrophy, pontobulbar palsy, sensorineural deafness. | Autosomal recessive | Brown Vialetto-Van Laere syndrome 2 |
| PCH1A | VRK1 | 602168 | Vaccinia related to serine/threonine kinase 1 | 14q32.2 | Encoding serine/threonine protein kinases with vaccinia | Early infantile. Microcephaly, severe hypotonia, areflexia, central visual impairment, dysphagia, respiratory failure. | Autosomal recessive | Pontocerebellar hypoplasia with infantile muscular atrophy |
| PCH1B | EXOSC3 | 606489 | Exosome component 3 | 9p13.2 | Encoding non-catalytic component 3 of the human exosome | Early infant. Microcephaly, mental retardation, early death. | Autosomal recessive | Pontocerebellar hypoplasia with infantile muscular atrophy |
| PCH1C | EXOSC8 | 607596 | Exosome component 8 | 13q13.3 | Encoding non-catalytic component 8 of the human exosome | Early infant. Microcephaly, mental retardation, early death. | Autosomal recessive | Pontocerebellar hypoplasia with infantile muscular atrophy |
| SMAPME | ASAH1 | 613468 | N-acylsphingosine amidohydrolase 1 | 8p22 | Encoding family of acid ceramidase proteins | Early childhood. Proximal muscle weakness, hypotonia, areflexia, muscular atrophy, fasciculations, sensorineural hearing loss, respiratory muscle weakness, microcephaly. | Autosomal recessive | Spinal Muscular Atrophy with Myoclonic Epilepsy |
| LAAHD | GLE1 | 603371 | RNA export mediator GLE1 | 9q34.11 | Coding polypeptide homologue with yeast Gle1p | Antenatal. Fetal immobility, hydrops, micrognathia, pulmonary hypoplasia, joint contractures. | Autosomal recessive | Lethal arthrogryposis with anterior hasta cell disease |
| SMARD1 | IGHMBP2 | 604320 | Immunoglobulin helicase binding to μ protein 2 | 11q13 | Encode helicase superfamily member | Early infantile. Distal and lower extremity muscle weakness, early diaphragmatic weakness. | Autosomal recessive | Spinal muscular atrophy with diaphragmatic paralysis |
| SMARD2 | LAS1L | 309585 | Lanosterol synthase 1 | Xq12 | Activates RNA binding activity | Early infantile. Distal and lower extremity muscle weakness, early diaphragmatic weakness. | X-linked recessive | Spinal muscular atrophy with diaphragmatic paralysis |
| DSMA | TRPV4 | 600175 | Transient receptor cationic potential transient receptor subfamily V member 4 | 12q24.11 | Encode Ca2-permeable nonselective cation channel family | Congenital. Muscle weakness, distal and proximal leg contractures, sensorineural hearing loss. | Autosomal dominant | Congenital spinal muscular atrophy of the lower extremities |
| SMALED1 | DYNC1H1 | 600112 | Cytoplasmic dynein 1 heavy chain 1 | 14q32.31 | Cytoplasmic dynein heavy chain family encoding | Congenital until adulthood. Proximal muscular deformity in legs respects adductors and semitendinosus, not progressive. | Autosomal dominant | Spinal muscular atrophy in lower extremities-1 |
| SMALED2 | BICD2 | 609797 | BICD charging adapter 2 | 9q22.31 | Negative end motility by dynein in microtubules | Congenital until adulthood. Slow progression, proximal muscle weakness in legs with some contractures. | Autosomal dominant | Spinal muscular atrophy in lower extremities-2 |
Source: Peeters K. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. Castiglionia C. Spinal muscular atrophies not associated with SMN1. Clinica Las Condes Medical Journal.
Table 2: Genotypic and phenotypic characteristics associated with the presentation of late-onset non-5q SMA.
| Type | Gene | OMIM | Name | Location chromosomal | Function | Clinic | Heritage | Condition |
|---|---|---|---|---|---|---|---|---|
| LATE START | ||||||||
| HMSNP | TFG | 602498 | Trafficking from endoplasmic reticulum to Golgi regulator | 3q12.2 | Encoding fusion oncoproteins | Young adults. Painful muscle paresthesias, myotonia in hands, dysphagia, muscle weakness, proximal or distal muscle atrophy, fasciculations, subsequent slowly progressive distal sensory impairment. | Autosomal dominant | Hereditary sensory motor neuropathy proximal, Okinawa type |
| LGMD1B | LMNA | 150330 | Lamin A/C | 1q22 | Encoding nuclear lamina protein | Young adults. Progressive proximal muscle weakness and cardiomyopathy. | Autosomal dominant | Adult-onset proximal spinal muscular atrophy followed by cardiac involvement. |
| SMAX1 | AR | Sin OMIM | Androgen receptor | Xq12 | Encoding androgen receptor | Adulthood. Proximal weakness, bulbar, endocrine disorder. | X-linked recessive | Kennedy's disease, X-linked bulbo-spinal atrophy. |
| SMAJ | CHCHD10 | 615903 | Domain containing 10 Coiled-Coil-Helix-Coiled-Coiled-Coil-Helix | 22q11.23 | Encoding mitochondrial proteins | Adulthood. Painful paresthesias, fasciculations, muscle weakness and areflexia. | Autosomal dominant | Spinal muscular atrophy type Jokela |
| ALS4 | SETX | 608465 | Senataxin | 9q34.13 | Sen1p protein coding | Juvenile or adult onset. Proximal and distal weakness, hand tremor, sharp reflexes. | Autosomal dominant | Juvenile or adult-onset spinal muscular atrophy with pyramidal features |
| SPSMA | TRPV4 | 605427 | Member 4 of subfamily V of transient receptor potential cation channels. | 12q24.11 | Encoding a member of the transient receptor potential channel subfamily. | Early adulthood. Progressive weakness of facial and pectoral muscles with laryngeal paralysis, sensorineural deafness, skeletal abnormalities preserving medial gastronecmium and biceps femoris. | Autosomal dominant | Scapulo-peroneal spinal muscular atrophy |
| SMAFK | VAPB | 605704 | Synaptobrevin-like protein B- and C-associated protein (VAMP). | 20q13.32 | Encoding type IV membrane protein | Adulthood. Paresthesias and muscular fasciculations. | Autosomal dominant | Spinal Muscular Atrophy of late onset Finkel's Type |
| SMAFK | HEXB | 606873 | Hexosaminidase beta subunit | 5q13.3 | Encoding Hexosaminidase beta subunit | late adult onset. Proximal muscle weakness of lower extremities | Autosomal recesive | Late adult-onset pure spinal muscular atrophy |
Source: Peeters K. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. Castiglionia C. Spinal muscular atrophies not associated with SMN1. Clinica Las Condes Medical Journal.
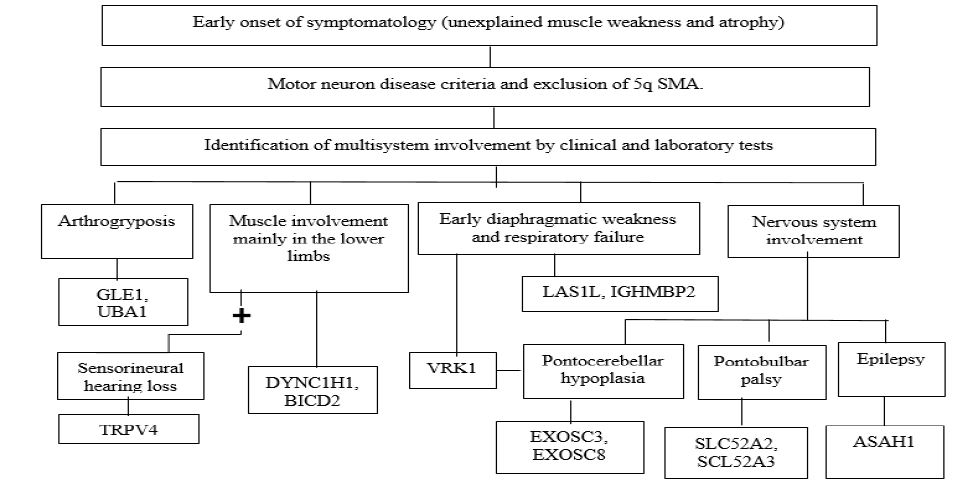
Source: Peeters K. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. Castiglionia C. Spinal muscular atrophies not associated with SMN1. Clinica Las Condes Medical Journal.
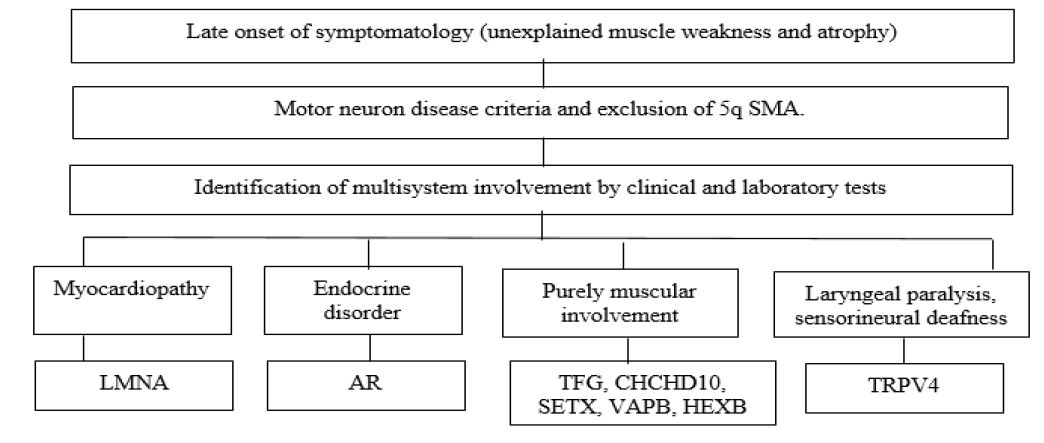
Source: Peeters K. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. Castiglionia C. Spinal muscular atrophies not associated with SMN1. Clinica Las Condes Medical Journal.
The follow-up of patients diagnosed with SMA in the presymptomatic stage requires monitoring of the development of symptomatology to determine the appropriate initiation of targeted or supportive therapies, the multidisciplinary evaluation should be carried out every six months to assess: respiratory and motor function, orthopedic status and nutritional status, this support is essential to reduce the severity of symptoms [23].
As it is a progressive neuromuscular disease, the functional prognosis, respiratory capacity and life expectancy will depend on the time of evolution of the disease, the multidisciplinary management in a timely manner and the establishment or not of complications among which are swallowing problems with subsequent affectation in nutrition; Gastrointestinal dysfunction with constipation, gastroesophageal reflux and delayed gastric emptying and respiratory problems consisting of airway obstruction and aspiration infections, in relation to the above, the prognosis of a patient with the presence of complications and without multidisciplinary interventions has a life expectancy rarely greater than 2 or 3 years [4,24].
Materials and methods
A 32-year-old female patient, with clinical picture of onset at the age of 1 year and 3 months with the presence of equinismus, varus, rearfoot supination, right forefoot adduction and bilateral wrist extension limitation, with subsequent onset of weakness and muscle atrophy in predominantly lower limbs, denying the presence of proprioceptive, auditory, visual and skin alterations. As background product of non-consanguineous parents, no family history of degenerative neuromuscular diseases.
On physical examination symmetrical limbs, symmetrical pulses, no distal coldness, capillary filling less than 2 seconds, fasciculations and pain on palpation at the level of the rhomboid muscle, bilateral hand posture, no hyperextension, no ligament hyperlaxity, with presence of splints in lower limbs, muscle atrophy, generalized areflexia, walks with support, strength according to Daniels scale with absence of contraction in flexor-extensors of hands and feet, positive Gowers sign, no sitting, neurologically alert, oriented in time, place and person.
With paraclinical tests showing Creatine kinase 239 IU/L, calcium 9.4 mg/dl, prolactin 19.14 ng/mL, Thyroid Stimulating Hormone (TSH) 5.04 mIU/L, parathyroid hormone (PTH) 51.91 pg/mL, vitamin D 25 ng/mL, 17-OH-Progesterone 19. 7 IU/l, Somatomedin C 181 U/ml, electrocardiogram, transthoracic echocardiogram and holter without cardiovascular alterations, orthoradiography of spine with right vertex rotoescoliosis at lumbar dorsal level with Cobb angle of 44 degrees, sural nerve biopsy with loss of axons with little demyelination, hypertrophy of Schwann cells, without electromyography.
According to the degenerative neuromuscular clinical manifestations and the results of the sural nerve biopsy, the first diagnostic impression was Charcot-Marie-Tooth disease, for which reason a molecular study of deletions/duplications in the PMP22 gene was performed, with the result that there was no alteration.
In view of the suspicion of a progressive neurodegenerative polyneuropathy to be classified and the wide phenotypic and genotypic variability associated with this medical condition, a trio clinical exome sequencing study was performed using Illumina technology from a peripheral venous blood sample; DNA extraction was carried out using Qiagen’s DNeasy package; to determine the concentration and purity of DNA, the samples were evaluated using a spectrophotometer (Na-noDrop), obtaining values of approximately 500 ng/uL and an average Optical Density (OD) A260/A280 of 1.80. Subsequently, massive sequencing of Nextera TM libraries was performed using the Illumina platform with 100X coverage. Alignment with reference genome GRCh38. All selected regions presented depth greater than or equal to 32.2 x and a minimum mapping confidence threshold Q30 with a total read of 27,320,632 Nexteratm Illumina libraries. The results were compiled in a Variant Call Format (VCF) output file, where the variants found were recorded. The population databases Exac, 1000 Genomes, OMIM and gnom AD were consulted to determine the existence of the variants reported; subsequently, bioinformatic analysis and functional effect prediction of the variants found were performed.
For the analysis of the reported variants we used the bioinformatics software: Mutation Taster (http://www.mutationtaster.org/), PROVEAN (http://provean.jcvi.org/index.php), the UMD predictor (http://umd-predictor.eu/), POLYPHEN (http://genetics.bwh.harvard.edu/pph2/) SIFT (https://sift.bii.a-star.edu.sg/), Human Splicing Finder (http://umd.be/Redirect.html) and Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), which served as in-situ clinical prediction tools. The nomenclature used to name the variants was based on the recommendations of the American College of Medical Genetics and Genomics (ACMG). Finally, a gene interaction network was performed using the GeneMania program to determine close associations with other genes to determine physical interactions or co-expression levels.
Results
In trio clinical exome sequencing a pathogenic variant was found in the DYNC2H1 gene with nucleotide change: c.8365 T>C, protein: p. Phe2789Leu, zygosity: homozygous, heterozygous father and mother; gene coding for cytoplasmic dyneins of heavy chain 1, subunit of the primary motor protein responsible for retrograde axonal transport in neurons which when affected is associated with the presentation of muscular affectation predominantly in the proximal lower extremity [25].
On the other hand, the second variant with clinical pathogenic significance was found in the trio clinical exome sequencing in the NOD2 gene; this variant was not identified in the DNA extracted from any of the parents, with nucleotide change: c.1001G>A, protein: p.Arg334Gln, zygosity: heterozygous, gene that plays an important role in the function of the immune system, however, the genetic variant has not been associated with any type of clinical manifestation [27].
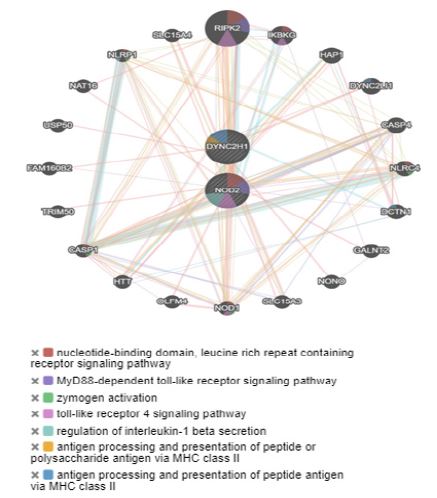
Additionally, in order to determine interactions between the affected genes to establish a phenotype-genotype relationship, a gene interaction network was constructed (Figure 3). In this network it was observed that the two genes involved in the clinical pathology of the patient, DYNC2H1 and NOD2, according to the analysis with GeneMania, presented close interactions with the genes RIPK2, NLRP1, NOD1, IKBKG, DYNC2LI1, NLRC4, DCTN1 and CASP1, all with process-related functions to the nucleotide-binding domain, leucine-rich repeat-containing receptor signaling pathway, MyD88-dependent Toll-like receptor signaling pathway, zymogen activation, Toll-like receptor 4 signaling pathway, regulation of interleukin-1 beta secretion, antigen processing, and presentation of peptide or polysaccharide antigens through Major Histocompatibility Complex (MCH) class II. The genes highlighted for their co-expression with DYNC2H1 and NOD2 genes were RIPK2, NOD1 and TNFAIP3.
The DYNC2H1 gene presented physical interaction with 6 different genes, having a high interaction with 1 of them (DYNC2LI1). The NOD2 gene was the gene that reported the most physical interactions [16], being the association with the RIPK2 gene the strongest interaction.
Knowing the metabolic pathways, interactions, allows to know the pathophysiological mechanisms involved and to establish a specific diagnosis in a degenerative and progressive neuromuscular disease with a phenotypic and genotypic variability as the non-5q SMA in order to offer a targeted treatment that manages to impact on the prognosis and morbidity and mortality attributed to this pathology, establishing follow-up guidelines, education on risk of heritability and an adequate genetic counseling approaching 4P medicine that would impact on the natural history of the disease [2,4,28].
Elaboration: Own source, Gene Mania Program.
Bioethical aspects
The present case report is considered an observational descriptive study based on the review of the clinical history of a patient and review of the literature; no intentional modification of the biological, physiological or psychological characteristics was made, which is why the level of this research has been categorized as minimal risk according to resolution 8430 of 1993. Prior to the production of the case report, the respective informed consent for the confidential use of data was obtained. The authors declare that in this article the data taken from the clinical history were analyzed protecting the confidentiality and privacy of the patient.
Discussion
In Colombia, SMA are considered orphan diseases that have coexisted in the population since their origin, however, they have gone unnoticed in relation to their low prevalence and lack of knowledge of health personnel that leads to the difficult diagnosis, as described by Castiglioni and Peeters et al. [9,15] 5q SMAs account for 80% of motor neuron disorders, however, the number of causative genes associated with non-5q SMAs, has expanded rapidly due to the advent of next generation sequencing technologies, existing according to Castiglioni and Lozano et al. [9] up to 2018, 20 associated genes, which is why obtaining a molecular study that reports non-pathogenic clinical significance in the SMN1 gene does not rule out the disease and more genomic data should be included for the characterization of the patient with suspected SMA.
Exome sequencing is aimed at obtaining the maximum possible genetic information from the exons present in the almost 26,000 genes of human beings, covering approximately 85% of the variants associated with the presentation of the heterogeneous group of complex hereditary genetic diseases, elucidating the diagnosis of the characteristic phenotype of the patient and the discovery of new genetic variants associated with pathologies [28].
Currently, as part of exome analysis targeting genes of interest, sequencing of the so-called clinical exome is in use, using kits developed by the industry (Agilent SureSelect Focused Exome, Illumina TruSight One, Roche NimbleGen SeqCap EZ MedExome), which include about 5,000 genes included in the OMIM (Online Mendelian Inheritance in Man) catalog and associated with clinically relevant disease-associated phenotypes. That is, they cover only 20% of the entire exome and require, like the gene panels, additional analysis if the alteration causing the clinical condition is not found, clinical use has been proposed as the first diagnostic test in neurodevelopmental disorders [28].
Clinical bioinformatics is defined as an emerging scientific discipline in the biomedical sciences that uses information technology to organize, analyze and distribute biological information, using DNA, RNA, amino acid sequences, proteins, three-dimensional molecular structures, gene interactions, metabolic pathways, among others; This has made it possible to explain the function of various proteins, develop structural models and protein interaction networks, elucidate the different molecular mechanisms related to the presentation of different diseases, improve the understanding of the relationship between heredity and the risk of suffering a disease, reveal the genetic influence on the appearance and identification of biomarkers and specific therapeutic strategies for these conditions [29].
Seeking to define a specific diagnosis according to the heterogeneous clinical manifestations of the patient, a clinical triple exome sequencing study was performed in search of genomic variants associated with the patient’s phenotype and to perform a correct correlation of the protein function by means of bioinformatics techniques. In this context, in the present study, 2 genetic variants with a pathogenic clinical significance were identified in the DYNC1H1 and NOD2 gene [28].
The DYNC1H1 gene is responsible for making the protein dynein-2, with subsequent development of dynein-2 complex which is found in cilia, microscopic projections that protrude from the cell surface, dynein-2 is involved in Intraflagellar Transport (IFT) by which materials are transported within the cilia; specifically, dynein-2 is a motor that uses energy from the ATP molecule to drive the transport of materials from the tip of the cilia to the base [25].
The DYNC1H1 and DYNC2H2 gene family encodes cytoplasmic dynein heavy chain 1, which is a subunit of the primary motor protein responsible for retrograde axonal transport in neurons; when a genetic variant with pathogenic clinical significance is present in this gene, these functions are altered [25].
According to studies performed by Weedon et al. [26] where a nonsense mutation [p.H306R (c.917A> G)] in the DYNC1H1 gene was identified in a family of four generations with 23 members, the most striking characteristic among the patients was a unique distribution of muscle involvement. The quadriceps femoris muscle was affected from the early course of the disease, and the proximal lower extremity was predominantly involved throughout the course of the disease.
Harms et al. [25]. meanwhile reported three nonsense variants in the tail of the DYNC1H1 domain in families with dominant spinal muscular atrophy with lower extremity predominance (SMA-LED, OMIM 158600), expanding the role of DYNC1H1 in motor neuron maintenance. In their work Harms et al. [25] proposed that the non-progressive clinical course of the disease despite early childhood onset should be another hallmark of SMA-LED; they further postulated that the DYNC1H1-associated gene family plays an essential role in maintenance of spinal motor neurons and their axons.
Tsurusaki et al. [10] studied two siblings: the first had mild motor delay in infancy, unsteady gait that persisted until 3 years of age, examinations showing proximal lower limb muscle atrophy and decreased deep tendon reflex; Gowers sign was positive. No neurological deficit was demonstrated. Motor nerve conduction velocity within normal limits. Muscle computed tomography demonstrated severe atrophy and lipid degeneration, predominantly in the bilateral quadriceps femoris muscle. A muscle biopsy of the quadriceps femoris muscle demonstrated severe atrophy of type 2 fiber bundling.
The second patient, presented with delayed motor development; physical examination revealed moderate proximal lower extremity muscle weakness, but Gowers’ sign was negative, muscle computed tomography revealed severe atrophy and lipid degeneration, mainly in the bilateral quadriceps femoris muscle. Proximal lower extremity weakness and wasting is evident, but the patient showed no upper extremity weakness. The investigators found nonsense variants in the DYNC1H1 and DYNC2H2 genes in the 2 patients [10].
Scoto et al. [21] describe the phenotype of patients with pathogenic clinical significance in the DYNC1H1 gene, where it is usually characterized by muscle weakness with predominance in lower limbs in proximal segments that relatively preserves adductor and semitendinosus muscles, muscle atrophy with absence of sensory alteration, anadine gait, gait delay and loss of distal osteotendinous reflexes.
On the other hand, the pathogenic variant in the NOD2 gene not identified in DNA extracted from leukocytes of either parent, formerly known as CARD15 according to Guzman et al. [22] manufactures a protein that plays an important role in the function of the immune system, is active in some types of immune system cells (including monocytes, macrophages and dendritic cells), which help protect the body against bacteria and viruses.
The NOD2 protein has several critical functions in the body’s defense against foreign invaders, it is involved in the recognition of certain bacteria and in stimulating the immune system to respond appropriately, when activated by specific substances produced by bacteria, the NOD2 protein activates a protein complex called nuclear factor-kappa-B, this protein complex regulates the activity of multiple genes, including genes that control immune responses and inflammatory reactions, however, despite presenting genetic variants it has not been associated with any type of clinical manifestation [27].
Conclusion
Non-5q SMA is a set of hereditary neurodegenerative disorders capable of causing an alteration in the anterior horn cells in the spinal cord and motor nuclei in the lower part of the brainstem and subsequently a high phenotypic and genotypic variability that generate an impact on quality of life, psychosocial, emotional and functional development; Although most patients are due to variants in the SMN1 gene, there are other non-5q genes associated with this pathology, which is why the importance of performing genomic studies, clinical bioinformatics, interaction networks and reverse phenotyping based on the selection of a group of people with a genetic variant and the evaluation of its phenotype, which have become fundamental tools for the characterization of these complex diseases and the determination with the potential to change reactive medicine into preventive medicine, achieving an early diagnosis, with the possibility of initiating an early and directed treatment that has an impact on the prognosis and morbimortality attributed to this pathology, establishing follow-up guidelines and adequate genetic counseling that allows education on the risk of inheritability or presentation of a disease, bringing us closer to precision medicine for the sake of the 4P medicine that would impact on the natural history of the disease [1,15,31].
Conflict of interest: The authors declare that they have no conflicts of interest.
References
- Torricelli RE, Erazo Torricelli R, Neuromuscular U. UPDATE ON TREATMENTS FOR SPINAL MUSCULAR ATROPHY [Internet]. Medicinabuenosaires.com [cited 2023 Jan 28]. Available from: https://www.medicinabuenosaires.com/revistas/vol82-22/s3/76s3.pdf
- Bodamer OA, Spinal muscular atrophy [Internet]. Uptodate. [cited 2023 Jan 28]. Available from: https://www.uptodate.com/contents/spinal-muscular-atrophy?search=atrofia%20muscular%20espinal&source=search_result&selectedTitle=1~68&usage_type=default&display_rank=1.
- Bolaño Díaz CF, Morosini M, Chloca F, Mesa L, Jáuregui A, Pirra L, et al. The difficult path to diagnosis of the patient with spinal muscular atrophy. Arch Argent Pediatr [Internet]. 2022; e202102542. DOI: https://doi.org/10.5546/aap.2021-02542.eng
- Castellano IP, Cabrera M, Calvo R. Delphi consensus of recommendations for the treatment of patients with spinal muscular atrophy in Spain (RET-AME consensus). Neurology [Internet]. 2021; p216-228. DOI: https://doi.org/10.1016/j.nrl.2021.07.008
- Madruga-Garrido M, Vázquez-Costa JF, Medina-Cantillo J, Brañas M, Cattinari MG, de Lemus M, et al. Design of a non-interventional study to validate a set of patient- and caregiver-oriented measurements to assess health outcomes in spinal muscular atrophy (SMA-TOOL study). Neurol Ther [Internet]. 2021; 10: 361-373. DOI: https://doi.org/10.1007/s40120-020-00229-w
- Cardona N, Ocampo SJ, Estrada JM, Mojica MI, Porras GL, et al. Clinical and functional characterization of patients with spinal muscular atrophy in central-western Colombia. Biomed. [Internet]. 2022; 42: 89-99. DOI: https://doi.org/10.7705/biomedica.6178.
- Instituto Nacional de Salud, Comportamiento de la notificación al Sivigila de las enfermedades huérfanas - raras, Colombia, 2021 hasta semana epidemiológica 20, [cited 2023 January 28, 2023]; Available from: https://www.ins.gov.co/buscador-eventos/BoletinEpidemiologico/2021_Boletin_epidemiologico_semana_24.pdf.
- Instituto Nacional de Salud, Proportion of notification of orphan - rare diseases, Colombia, 2016 until epidemiological period I of 2021, [cited 2023 January 28, 2023]; Available from: https://www.ins.gov.co/Biblioteca Digital/informe-de-evento-enfermedades-huerfanas-raras-2020.pdf#search=hiperlipoproteinemia
- Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. [Internet]. 2014; 137: 2879-2896.
- Tsurusaki Y, Saitoh S, Tomizawa K, Sudo A, Asahina N, Shiraishi H, Matsumoto NA. DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics. [Internet] 2012; 13: 327-332.
- Darras BT. Non-5q spinal muscular atrophies the alphanumeric soup thickens. Neurology. [Internet]. 2011; 42: 89-99.
- Chaytow H, Huang YT, Gillingwater TH, Faller KME. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol Life Sci [Internet]. 2018; 75: 3877-3894.
- Martínez-González JP, Guerrero-Vara R, Medina-Iglesias V. DYNC1H1 de novo mutation, spinal muscular atrophy and attention problems. Neurology. [Internet]. 2022; 37: 406-409.
- Giménez G, Prado F, Bersano C, Kakisu P. RESPIRATORY CARE OF PATIENTS WITH SPINAL MUSCULAR ATROPHY. Pediatric Pulmonology. [Internet]. 2022; 16: 23-29.
- Castiglionia C, Lozano-Arango A. Spinal muscular atrophies not associated with SMN1. Clinica Las Condes Medical Journal. [Internet]. 2018; 29: 643-653.
- Aguilar Carolina, Sepúlveda Rodrigo Andrés, Tagle Rodrigo. Stress ketoacidosis: Clinical case in patient with spinal muscular atrophy. Rev. med. Chile [Internet]. 2020; 148: 875-880. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872020000600875&lng=es. http://dx.doi.org/10.4067/S0034-98872020000600875.
- Specified Drug-Use Survey of Leuprorelin Acetate Injection Kit 11.25 mg “All-Case Investigation: Spinal and Bulbar Muscular Atrophy (SBMA)” [Internet]. Clinicaltrials.gov. [cited 2023 Jan 28]. Available from: https://clinicaltrials.gov/ct2/show/NCT03555578?term=leuprorelina&cond=Spinal+Muscular+Atrophy&draw=2&rank=2.
- Quilagüy Camelo N, León Chávez AF, Berrío Julio MP. Clinical aspects of Kennedy’s disease: Review Article. Ciencia Latina Revista Científica Multidisciplinar. Mexico [Internet]. 2022; 6: 898-913.
- Gene therapy for IGHMBP2-related diseases [Internet]. Clinicaltrials.gov. [cited 2023 Jan 28]. Available from: https://clinicaltrials.gov/ct2/show/NCT05152823?cond=IGHMBP2&draw=2&rank=1
- Lee J, Termglinchan V, Diecke S, Itzhaki I, Lam CK, Garg P, Lau E, et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature. [Internet]. 2019; 572: 335-340.
- Scoto M, Rossor AM, Harms MB, Cirak S, Calissano M, Robb S, et al. New mutations broaden the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology. [Internet]. 2015; 84: 668-679.
- Rivera EG, Patnaik A, Salvemini J, Jain S, Lee K, Lozeau D, Yao Q, et al. SARS-CoV-2/COVID-19 and its relationship to NOD2 and ubiquitination. Clin Immunol [Internet]. 2022; 238: 109027.
- Keinath MC, Prior DE, Prior TW. Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. Appl Clin Genet. [Internet]. 2021; 14: 11-25.
- Febrer A, Meléndez M. Spinal muscular atrophy. Complications and rehabilitation. Rehabilitation (Madr). [Internet]. 2001; 35: 307-311.
- Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology [Internet]. 2012; 78: 1714-1720.
- Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot- Marie-Tooth disease. Am J Hum Genet. [Internet]. 2011; 89: 308-312.
- Qingping Yao, Jean Schils, Distal lower extremity swelling as a prominent phenotype of NOD2-associated autoinflammatory disease, Rheumatology. [Internet]. 2013; 52: 2095-2097.
- Rubio S, Pacheco-Orozco RA, Gomez AM, Perdomo S, Garcia-Robles R. Next-generation sequencing (NGS) of DNA: present and future in clinical practice. Univ Med [Internet]. 2020; 61.
- Lago Sampedro AMa, García-Escobar E. Importance of bioinformatics in nowadays medicine. Is it really necessary in clinical practice? Nutr Hosp [Internet]. 2022; 39: 487-488. Disponible en: https://scielo.isciii.es/pdf/nh/v39n3/0212-1611-nh-39-3-487.pdf
- Instituto Nacional de Salud, Proportion of notification of orphan - rare diseases, Colombia, 2022 to epidemiological period VI, [cited 2023 Jan 28]; Available from: https://www.ins.gov.co/buscador-eventos/Informesdeevento/ENFERMEDADES%20HUERFANAS%20PE%20VI%202022.pdf
- From genotype to phenotype: Research strategy [Internet]. Genotyping. 2023 [cited 2023 Jan 28, 2023]. Available from: https://genotipia.com/genetica_medica_news/la-investigacion-del-genotipo-al-fenotipo/?utm_source=NL%20Genotipia%202022&utm_medium=email&utm_campaign=Genotipia%20Hoy%2024%20Enero%202023%20%2801GQF8CP5R7YVKH13DJ59% 29&_kx=G5f4ha6iy2_--vCEfJCsxoLcryft97m8xPNZaUcnw7JkGMjlFPUsT_m42645P7OC.SLZegA.